seqR
 seqR copied to clipboard
seqR copied to clipboard
fast and comprehensive k-mer counting package
seqR - fast and comprehensive k-mer counting package
![]()
![]()
![]()

About
seqR is an R package for fast k-mer counting. It provides
- highly optimized (the core algorithm is written in C++)
- in-memory
- probabilistic (with configurable dimensionality of a hash value used for storing k-mers internally),
-
multi-threaded (with a configurable size of the batch of
sequences (
batch_size) to process in a single step. Ifbatch_sizeequals 1, the multi-threaded mode is disabled, which potentially causes a longer computation time)
implementation that supports
- various variants of k-mers (contiguous, gapped, and positional counterparts)
- all biological sequences (e.g., nucleic acids and proteins)
Moreover, the result optimizes memory consumption by the application of sparse matrices (see package Matrix), compatible with machine learning packages such as ranger and xgboost.
How to…
How to install
To install seqR from CRAN:
install.packages("seqR")
Alternatively, if you want to use the latest development version:
# install.packages("devtools")
devtools::install_github("slowikj/seqR")
How to use
The package provides two functions that facilitate k-mer counting
-
count_kmers(used for counting k-mers of one type) -
count_multimers(a wrapper ofcount_kmers, used for counting k-mers of many types in a single invocation of the function)
and one function used for custom processing of k-mer matrices:
-
rbind_columnwise(a helper function used for merging several k-mer matrices that do not have same sets of columns)
To learn more, see features overview vignette and reference.
Examples
counting 5-mers
count_kmers(sequences=c("AAAAAVVAVFF", "DFGSADFGSA"),
k=5)
#> 2 x 12 sparse Matrix of class "dgCMatrix"
#> [[ suppressing 12 column names 'A.A.A.A.A_0.0.0.0', 'A.V.V.A.V_0.0.0.0', 'V.V.A.V.F_0.0.0.0' ... ]]
#>
#> [1,] 1 1 1 1 1 1 1 . . . . .
#> [2,] . . . . . . . 1 1 1 2 1
counting gapped 5-mers with gaps (0, 1, 0, 2) (XX_XX__X)
count_kmers(sequences=c("AAAAAVVAVFF", "DFGSADFGSA"),
kmer_gaps=c(0, 1, 0, 2))
#> 2 x 7 sparse Matrix of class "dgCMatrix"
#> A.A.A.A.A_0.1.0.2 A.A.V.V.F_0.1.0.2 A.A.V.A.F_0.1.0.2 A.A.A.V.V_0.1.0.2
#> [1,] 1 1 1 1
#> [2,] . . . .
#> G.S.D.F.A_0.1.0.2 F.G.A.D.S_0.1.0.2 D.F.S.A.G_0.1.0.2
#> [1,] . . .
#> [2,] 1 1 1
counting 1-mers and 2-mers
data(CsgA)
CsgA[1L:2]
#> $`sp|P28307|CSGA_ECOLI Major curlin subunit OS=Escherichia coli (strain K12) OX=83333 GN=csgA PE=1 SV=3`
#> [1] "M" "K" "L" "L" "K" "V" "A" "A" "I" "A" "A" "I" "V" "F" "S" "G" "S" "A"
#> [19] "L" "A" "G" "V" "V" "P" "Q" "Y" "G" "G" "G" "G" "N" "H" "G" "G" "G" "G"
#> [37] "N" "N" "S" "G" "P" "N" "S" "E" "L" "N" "I" "Y" "Q" "Y" "G" "G" "G" "N"
#> [55] "S" "A" "L" "A" "L" "Q" "T" "D" "A" "R" "N" "S" "D" "L" "T" "I" "T" "Q"
#> [73] "H" "G" "G" "G" "N" "G" "A" "D" "V" "G" "Q" "G" "S" "D" "D" "S" "S" "I"
#> [91] "D" "L" "T" "Q" "R" "G" "F" "G" "N" "S" "A" "T" "L" "D" "Q" "W" "N" "G"
#> [109] "K" "N" "S" "E" "M" "T" "V" "K" "Q" "F" "G" "G" "G" "N" "G" "A" "A" "V"
#> [127] "D" "Q" "T" "A" "S" "N" "S" "S" "V" "N" "V" "T" "Q" "V" "G" "F" "G" "N"
#> [145] "N" "A" "T" "A" "H" "Q" "Y"
#>
#> $`sp|P0A1E7|CSGA_SALEN Major curlin subunit OS=Salmonella enteritidis OX=149539 GN=csgA PE=1 SV=1`
#> [1] "M" "K" "L" "L" "K" "V" "A" "A" "F" "A" "A" "I" "V" "V" "S" "G" "S" "A"
#> [19] "L" "A" "G" "V" "V" "P" "Q" "W" "G" "G" "G" "G" "N" "H" "N" "G" "G" "G"
#> [37] "N" "S" "S" "G" "P" "D" "S" "T" "L" "S" "I" "Y" "Q" "Y" "G" "S" "A" "N"
#> [55] "A" "A" "L" "A" "L" "Q" "S" "D" "A" "R" "K" "S" "E" "T" "T" "I" "T" "Q"
#> [73] "S" "G" "Y" "G" "N" "G" "A" "D" "V" "G" "Q" "G" "A" "D" "N" "S" "T" "I"
#> [91] "E" "L" "T" "Q" "N" "G" "F" "R" "N" "N" "A" "T" "I" "D" "Q" "W" "N" "A"
#> [109] "K" "N" "S" "D" "I" "T" "V" "G" "Q" "Y" "G" "G" "N" "N" "A" "A" "L" "V"
#> [127] "N" "Q" "T" "A" "S" "D" "S" "S" "V" "M" "V" "R" "Q" "V" "G" "F" "G" "N"
#> [145] "N" "A" "T" "A" "N" "Q" "Y"
count_multimers(sequences=CsgA,
k_vector = c(1, 2))
#> 5 x 144 sparse Matrix of class "dgCMatrix"
#> [[ suppressing 144 column names 'R', 'L', 'Y' ... ]]
#>
#> [1,] 2 9 4 1 8 5 10 2 2 4 16 29 4 11 9 2 16 3 14 1 2 1 1 1 1 2 1 1 1 1 1 3 1 2
#> [2,] 3 8 5 2 7 6 11 2 2 4 17 22 3 11 10 2 20 1 15 1 3 1 . . 3 1 1 1 2 5 2 3 . 2
#> [3,] 3 8 5 2 7 6 11 2 2 4 17 22 3 11 10 2 20 1 15 1 3 1 . . 3 1 1 1 2 5 2 3 . 2
#> [4,] 2 9 4 1 9 5 10 2 1 4 15 30 4 11 9 2 17 4 13 1 2 1 1 1 1 2 1 1 1 1 1 3 1 2
#> [5,] 3 8 5 2 7 6 11 2 2 4 17 22 3 11 10 2 20 1 15 1 3 1 . . 3 1 1 1 2 5 2 3 . 2
#>
#> [1,] 1 3 1 3 1 2 7 1 1 1 1 3 1 1 2 2 1 1 12 3 1 2 1 1 1 1 1 1 1 1 1 1 1 2 1 2 2
#> [2,] . 2 . 2 1 2 3 . 1 2 . 3 1 . 3 2 2 1 6 4 2 3 1 2 1 . 1 . 1 1 . 1 . 1 1 2 .
#> [3,] . 2 . 2 1 2 3 . 1 2 . 3 1 . 3 2 2 1 6 4 2 3 1 2 1 . 1 . 1 1 . 1 . 1 1 2 .
#> [4,] 1 3 1 3 1 2 6 1 1 2 1 3 2 1 2 2 1 . 13 3 2 2 1 1 1 1 1 1 1 1 1 1 1 1 1 2 2
#> [5,] . 2 . 2 1 2 3 . 1 2 . 3 1 . 3 2 2 1 6 4 2 3 1 2 1 . 1 . 1 1 . 1 . 1 1 2 .
#>
#> [1,] 1 1 1 1 1 1 2 7 1 1 2 1 1 1 2 1 1 1 1 1 1 2 2 3 1 1 1 1 1 1 1 2 2 1 1 3 1
#> [2,] . 1 . 1 1 1 1 5 . . 2 . . 1 1 1 . . . 1 1 2 1 4 1 1 1 1 . 1 . 2 3 1 1 1 .
#> [3,] . 1 . 1 1 1 1 5 . . 2 . . 1 1 1 . . . 1 1 2 1 4 1 1 1 1 . 1 . 2 3 1 1 1 .
#> [4,] 1 1 . 1 1 1 1 7 1 1 2 1 1 1 2 1 . 1 1 1 1 2 2 3 1 . 1 1 1 1 1 1 2 1 1 3 1
#> [5,] . 1 . 1 1 1 1 5 . . 2 . . 1 1 1 . . . 1 1 2 1 4 1 1 1 1 . 1 . 2 3 1 1 1 .
#>
#> [1,] 1 1 2 1 2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
#> [2,] . 1 . 1 3 1 1 1 1 1 1 1 1 1 1 2 1 1 2 2 1 1 1 2 1 1 1 1 1 1 1 1 . . . .
#> [3,] . 1 . 1 3 1 1 1 1 1 1 1 1 1 1 2 1 1 2 2 1 1 1 2 1 1 1 1 1 1 1 1 . . . .
#> [4,] 1 1 2 1 2 . . . . . . . . . . . . . 1 . . . . . . . . . . . . . 1 1 1 1
#> [5,] . 1 . 1 3 1 1 1 1 1 1 1 1 1 1 2 1 1 2 2 1 1 1 2 1 1 1 1 1 1 1 1 . . . .
How to cite
For citation type:
citation("seqR")
or use:
Jadwiga Słowik and Michał Burdukiewicz (2021). seqR: fast and comprehensive k-mer counting package. R package version 1.0.0.
Benchmarks
The seqR package has been compared with other existing k-mer counting
R packages: biogram,
kmer,
seqinr, and
biostrings.
All benchmark experiments have been performed using Intel Core i7-6700HQ 2.60GHz 8 cores, using the microbenchmark R package.
Contiguous k-mers
Changing k
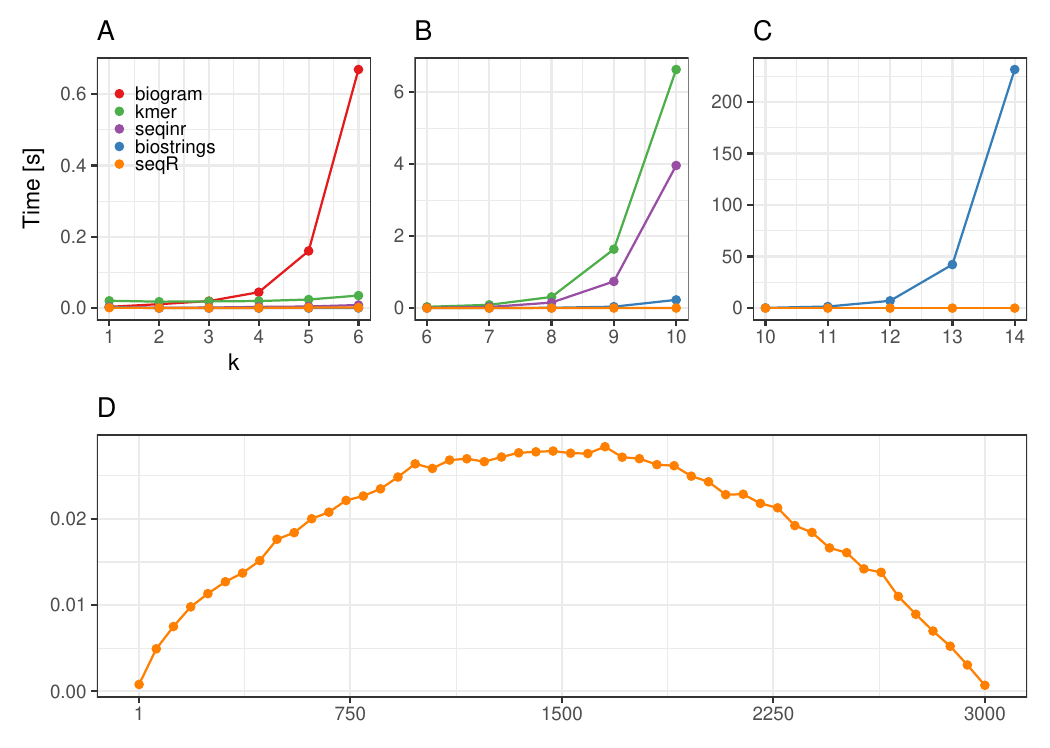
The input consists of one DNA sequence of length 3 000.
Changing the number of sequences
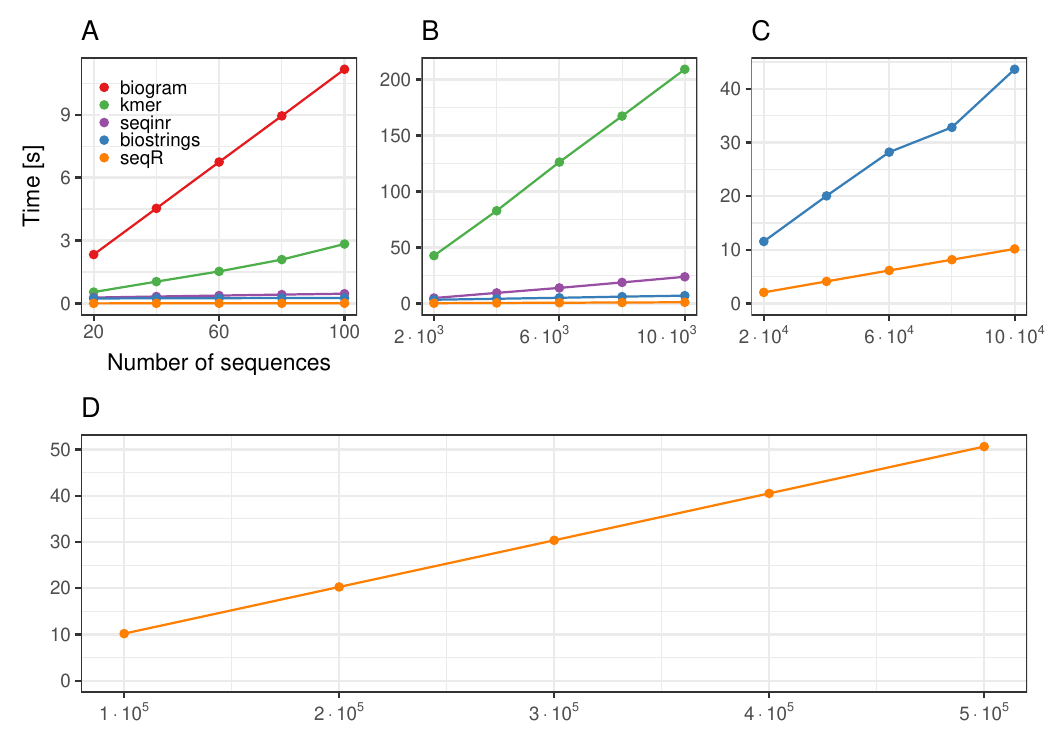
Each DNA sequence has 3 000 elements, contiguous 5-mer counting.
Gapped k-mers
Changing the first contiguous part of a k-mer
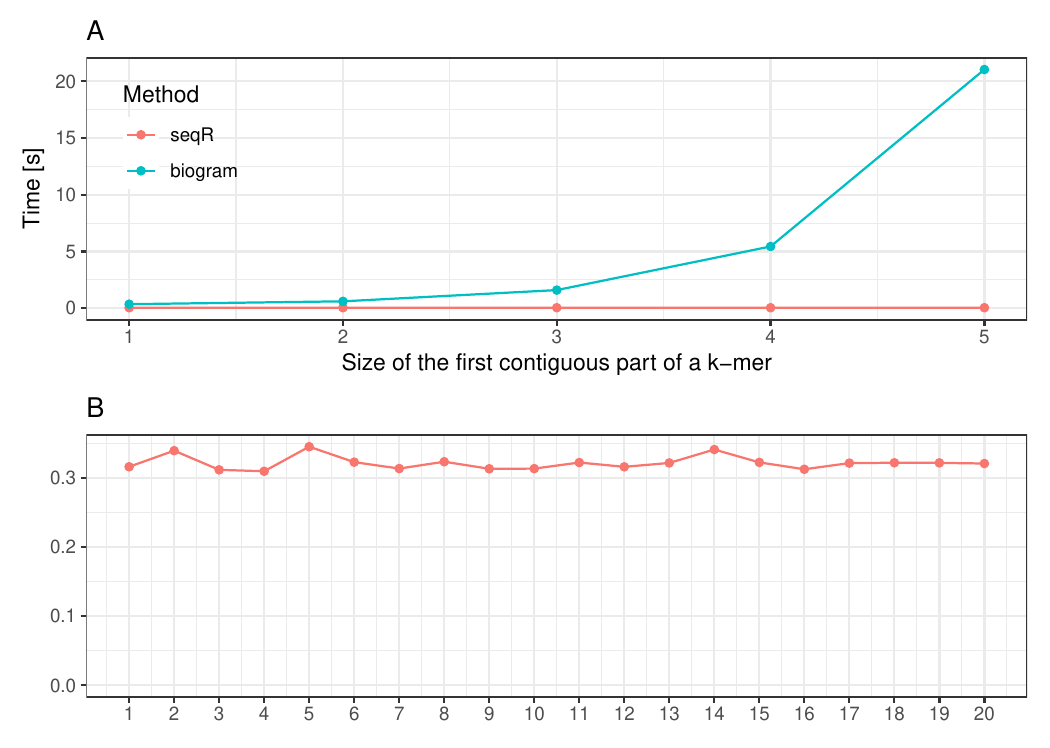
The input consists of one DNA sequence of length 1 000 000.
Gapped 5-mers counting with base gaps (1, 0, 0, 1).
Changing the first gap size
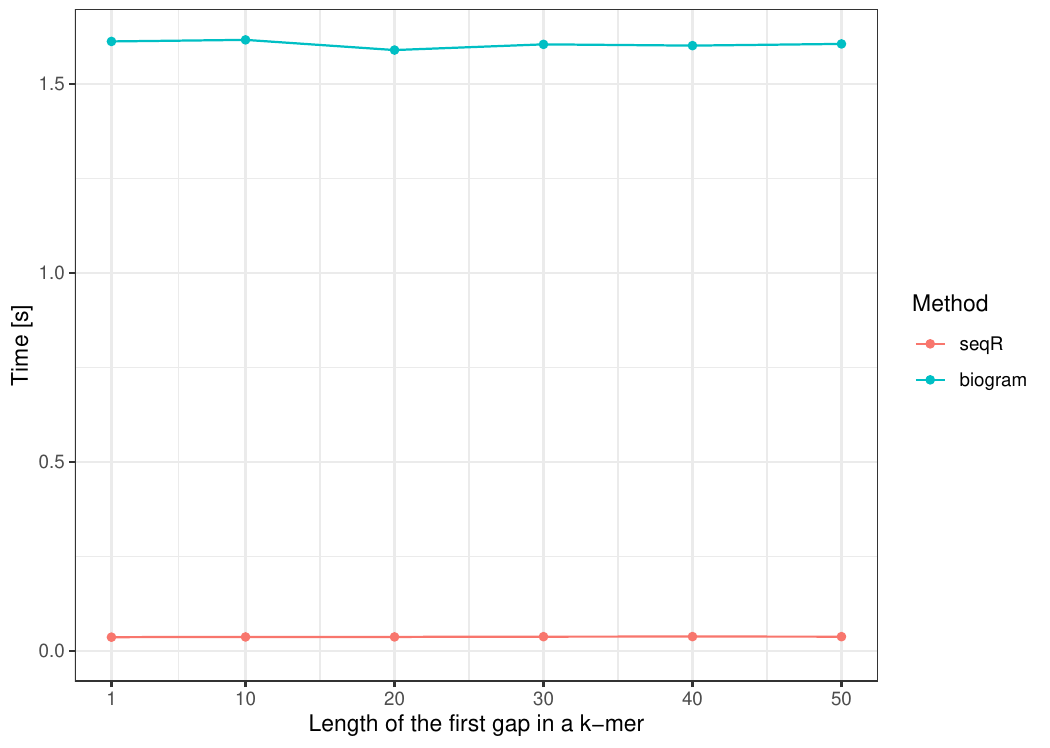
The input consists of one DNA sequence of length 100 000.
Gapped 5-mers counting with base gaps (1, 0, 0, 1).