Harshil Patel
Harshil Patel
Unable to replicate @JSchoenbachler 😏 I tried on a full-sized dataset on Nextflow Tower using the options below and it worked: `--genome GRCh37 --skip_alignment --pseudo_aligner salmon -r dev` 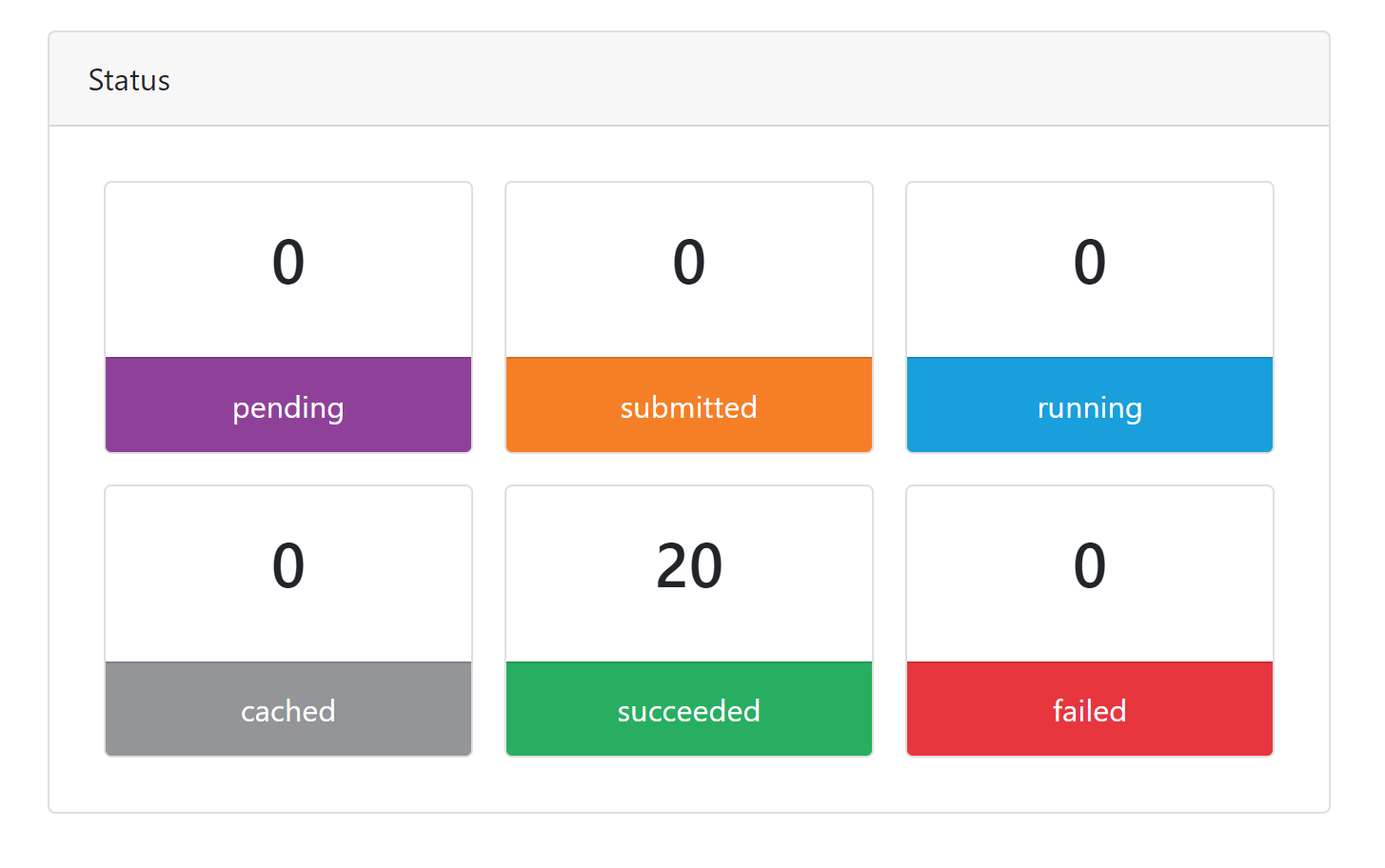 I...
Are you able to send me the `.command.out` and `.command.err` files in the `work` directory for the failing process? (and the `.nextflow.log` if you have it handy)? If it's sample...
Awesome. Thanks I have uploaded them here by adding a `.txt` extension. Sorry, didn't get time to do any more tests but I have just released v3.5. Will keep this...
Possibly looks like something we can add. Looks like it is being maintained [here](https://github.com/getzlab/rnaseqc). As mentioned in the [usage section](https://github.com/getzlab/rnaseqc#usage) we will need to always collapse the GTF file beforehand...
Thanks @noirot 👍 That has been fixed in the `dev` version of the pipeline as you can see [here](https://github.com/nf-core/rnaseq/blob/9745089ecea67db3b8a6a3014b01fc066776150f/main.nf#L1939). I think this issue is more related to running MultiQC on...
Should probably move this issue to MultiQC GitHub @ewels?
This will be fixed in MultiQC v1.12 https://github.com/ewels/MultiQC/issues/1484
MultiQC v1.12 was released recently but I found an issue with the rendering of the custom content of dupRadar that needs to be addressed before we bump versions and fix...
Fixed in https://github.com/nf-core/rnaseq/pull/874/commits/77d49ddeea097599601fb5253aface07d63839f7
Adding links where appropriate would definitely be a good start. Yep, it's not trivial to do this sort of thing without maybe having a bad dataset to compare to..