Sanity check BioType counts output
In issue #420 we had a tricky problem where the featureCounts BioType was creating a biotype entry for every gene. This worked fine, but then caused MultiQC to crash / time-out when it tried to make a bar plot with so many categories.
It would be good to add a simple wc -l check to the BioTypes count to check that there aren't more than about 100 lines or so in the output. If so, we delete the file so it doesn't get passed on.
A more elegant way to do it would be to do the check in the groovy code, then we could filter the channel and throw warnings to the console. But this will be more fiddly to write.
To be noticed, in version v1.4.2, if you use --skipBiotypeQC multiqc is not launched as line 1666 do not have .ifEmpty([])
Thanks @noirot 👍 That has been fixed in the dev version of the pipeline as you can see here. I think this issue is more related to running MultiQC on larger datasets.
Apologies to necro this thread, but would something like this help?
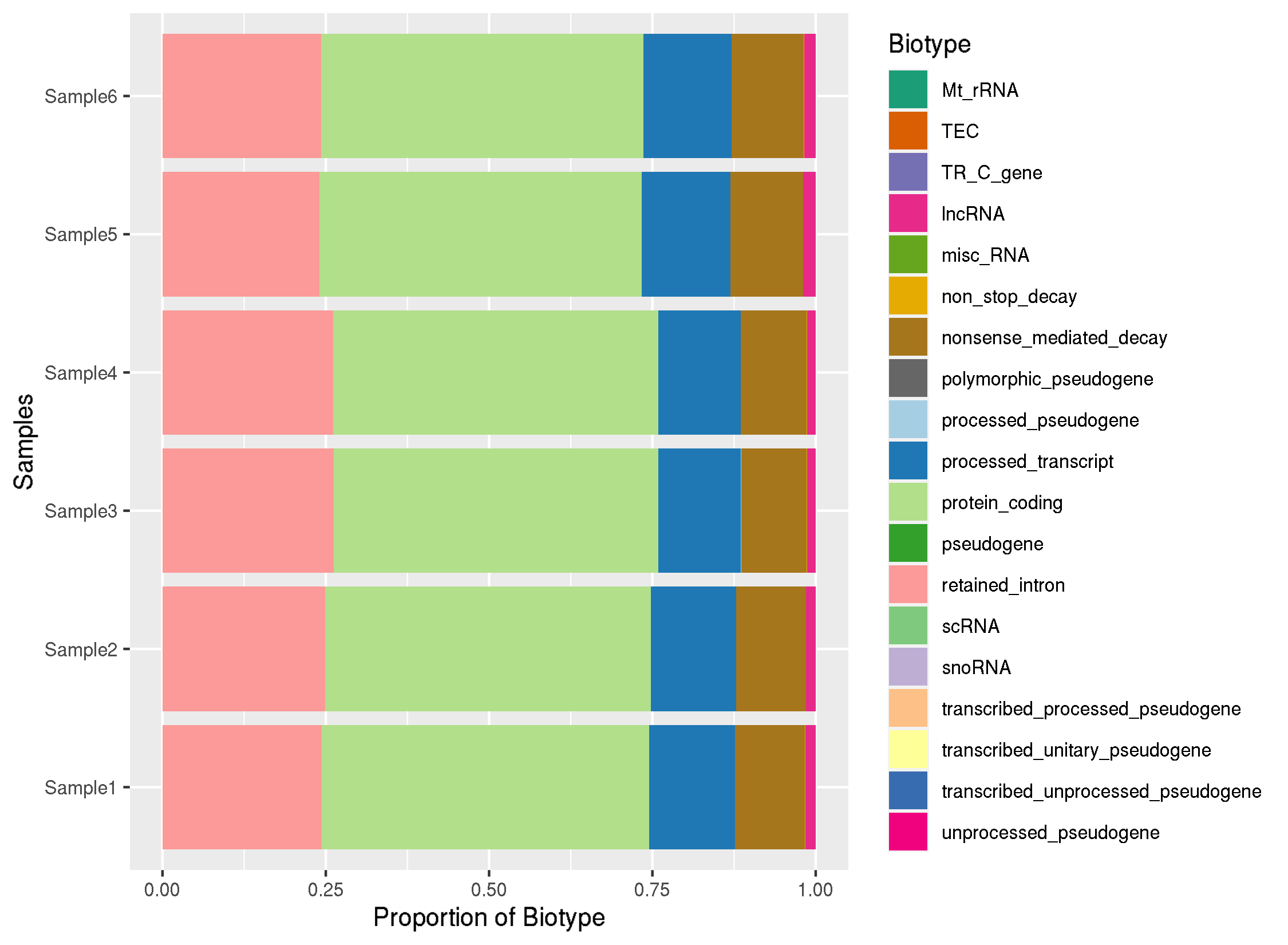
I'm using R to generate the Biotype plot, but I've generally observed that for Ensembl annotated Animal genomes they have like 40-50 biotypes, so some semblance of filter to remove features with very low reads assigned would be good start.
For getting this counts matrix, I'm using featureCounts, but alternatively I've used biomaRt to get biotype assignments in case I used salmon to quantify.
library(ggplot2)
library(reshape2)
data<- read.table("features.txt", sep="\t", header=T, row.names=1)
dim(data)
data <- data[rowSums(data) >= 1000,]
dim(data)
molten<- melt(as.matrix(data), id.vars="Biotype")
png("Biotype.png", res=300, units="in", height=5.5, width=7.5)
ggplot(molten, aes(x=Var2, y=value, fill=Var1)) + geom_col(position="fill")+
scale_fill_manual(values=c(RColorBrewer::brewer.pal(8,'Dark2'),RColorBrewer::brewer.pal(5,'Paired'), RColorBrewer
::brewer.pal(7,'Accent')))+
labs(x="Samples", y="Proportion of Biotype", fill="Biotype")+ coord_flip()
dev.off()
Which should generate images like this:
Some biotypes can then be clubbed together using Sequence ontology as well to remove terms if they're redundant.