wangshun1121
wangshun1121
@sfchen 相同的需求,一年半以后仍然存在!陈博士可否考虑规划一下这个功能?
Aha, you shouldn't try this repo this way, because in [NAMESPACE](https://github.com/immunomind/immunarch/blob/master/NAMESPACE) file of your package, `has_class` function was not exported, so it is only an internal function within your package!...
Maybe you can solve this issue yourself. I develop my Shannon function according to other similar functions. You can try to add sample names yourself?
Aha, I have similar demand, even though my CNV caller are cnv_facets and Sequenza rather than CNVkit. I wonder if maftools can accept CNV results from sources other than GISTIC.
[Facets.zip](https://github.com/PoisonAlien/maftools/files/7030173/Facets.zip) This is an example of Facets results. The most important file is `*.vcf.gz`, which gave details of every CNV region. By the way, definition of CNV types can be...
[Sequnza.zip](https://github.com/PoisonAlien/maftools/files/7030200/Sequnza.zip) Here are Sequenza outputs. In this folder, `*_segments.txt` is most import, CNV regions' details are recorded.
These two CNV callers are allele-specific, meaning that they can detect LOH regions without alteration of reads coverage: that's the advantage over callers depend only on coverage such as CNV-kit,...
Both, if possible
> @ctsa I tried to follow your suggestion #59 to use --forcedGT to re-genotyping tumor sample on merged calls. I check the log, it reports "Workflow successfully completed all tasks"....
@sangtaekim Well, this time I tried another bam file from white cell sequencing. The coverage of the site on normal is 37x, as is shown in following IGV screenshot: 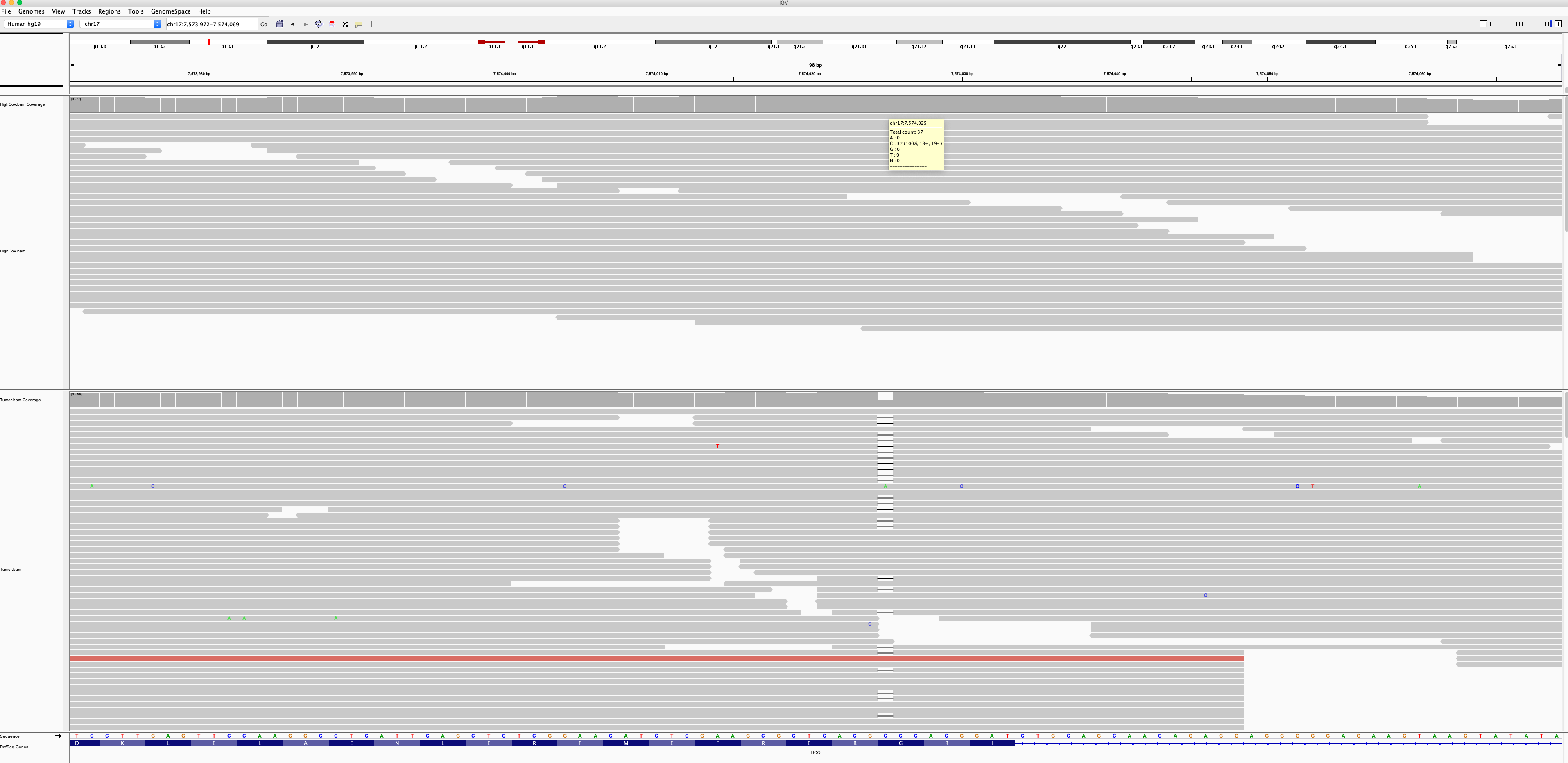...