Siebren Frölich
![]()
![]()
Siebren Frölich
Hi, you're welcome, and thanks for the tip! It looks like Rich extends on colorama, which we're already using. I'll have a look when I have the time!
Hey David, There is! `genomepy install -p local /path/to/local/directory/genomefile.fa.gz` Check out `genomepy install --help` for additional details (e.g. how to also install your own GTF)
Most install settings are universal, but there is one option specifically for local annotations: 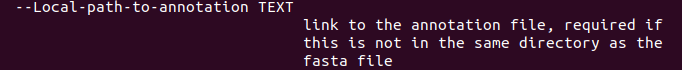 There is currently no additional documentation for the local provider. You're the second person who...
No. Unfortunately, making a generic SQL provider is a bit more difficult as there are at least 2 SQL variants (UCSC is using MariaDB for instance). I also don't see...
(As far as I know) you cannot do this with an empty samples.tsv, because snakemake creates its DAG by connecting rules to get from your `rule all` to your input...
if it works for rna-seq too we can throw out the old batch. they are too similar for differential gene expression anyway
Two options: - while fixing #471 I learned that `set()` sorts the data alphabetically. Should this be of influence here, you can use `seq2science.utils.unique()` instead. - you could perhaps prefix...
`-r -k --snakemakeOptions untill=[]` are fine. `--snakemakeOptions omit_from=[]` prompts a rerun.
these rules use threads in their params: - bam2cram - fastp_SE - fastp_PE - samtools_presort - samtools_sort these rules seem to get params changes listed: - multiqc_explain - multiqc_header_info -...
This happens with immature genomes/annotations. I suggest sticking with the older assembly for a while longer, but we could make the function "skip groups with errors" using flag `-allErrors`. What...