Colin Diesh
Colin Diesh
yes, storing multi-sample assembly-to-assembly alignment in BAM is another way. I am not a huge fan of storing in BAM, because it runs into trouble where you have to potentially...
sure thing, i'd be happy if you want to send that!
possible reference https://webpack.js.org/api/module-variables/#__webpack_chunk_load__-webpack-specific another example for vite
at least one other way i've seen this encoded "in the wild" is the 1000 genomes SV VCF which adds "RD_CN" to the genotypes field https://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000G_2504_high_coverage/working/20210124.SV_Illumina_Integration/1KGP_3202.Illumina_ensemble_callset.freeze_V1.vcf.gz
(it would be nice if there were somewhat of a standard as downstream tools can improve plotting capabilities on a larger set of VCF files that way :))
sort of similar idea in #4872 but different manifestation
Example: CNV track can help clarify what might be going on with coverage in a region from https://www.biorxiv.org/content/10.1101/2020.02.06.935742v1.full.pdf 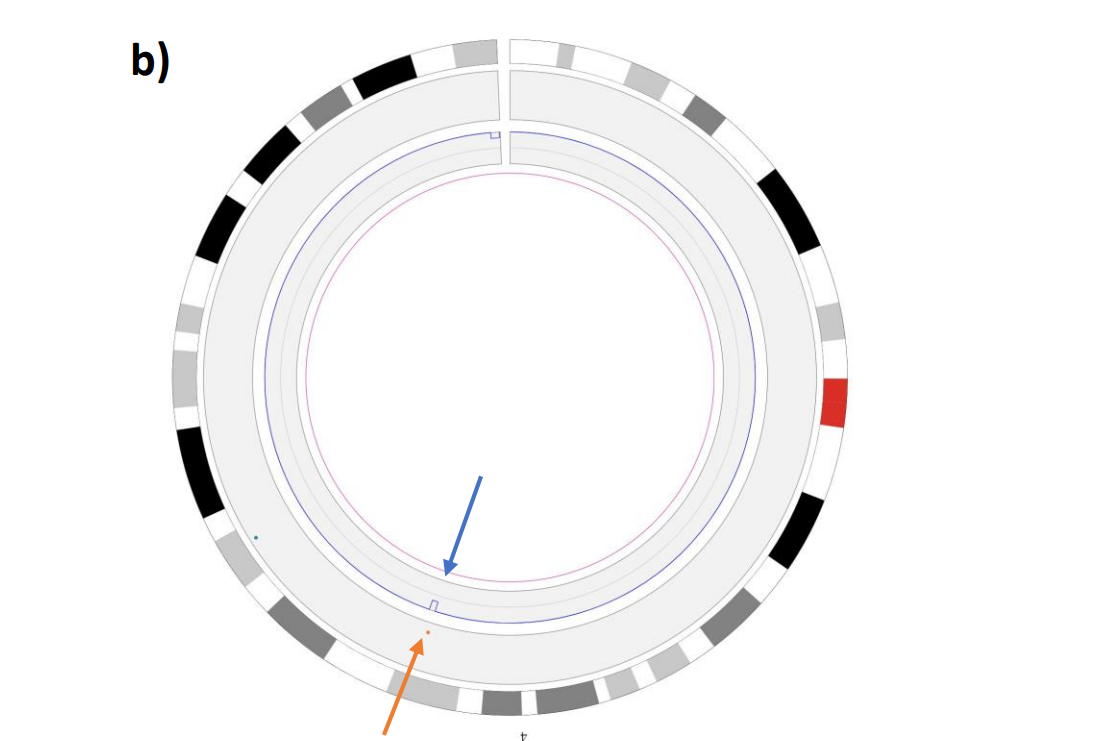 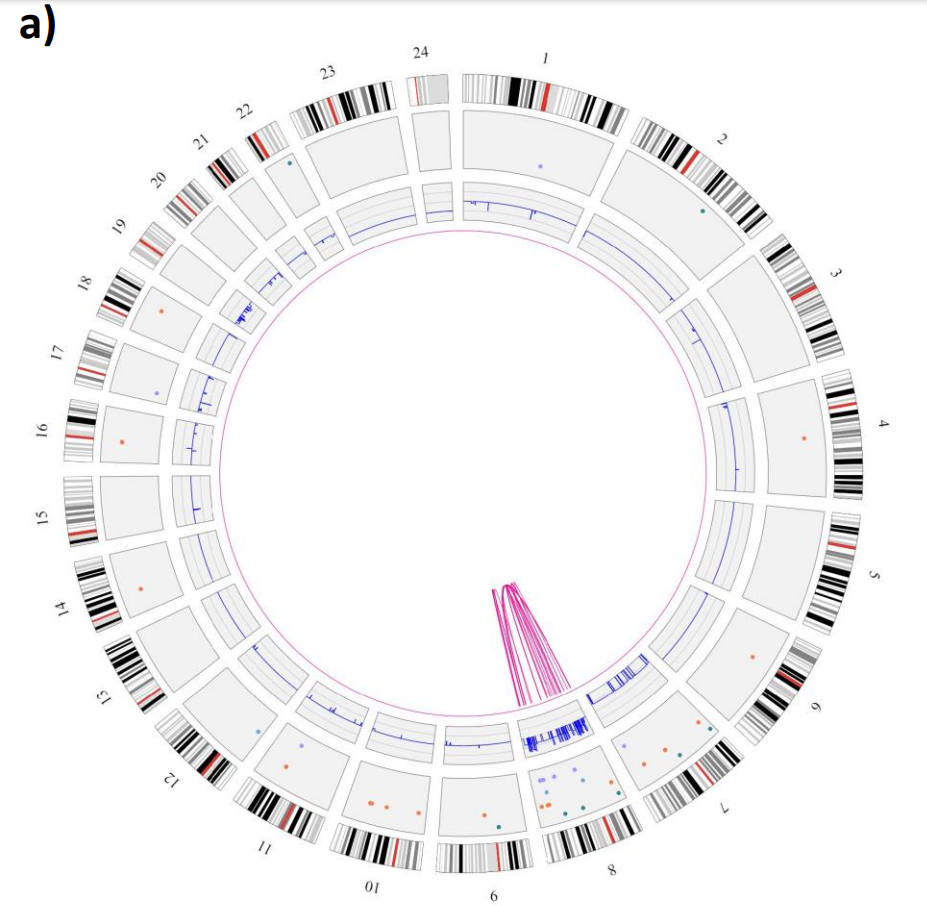
Another example showing the all-vs-all PAF (top) versus the multi-sample VCF (bottom) of the same region 
There could be a need for a 'multi-lgv synteny display', this would put each alignment data in it's own 'row'