ChenDepp
ChenDepp
hi, everyone! i hava a question about reference genome index file constructed by minimap2,Is there a detailed document? Similar to sam file format document? looking forward your reply, thanks
hi, everyone i running smudgeplot for my data, get strange smudgeplot. look the below 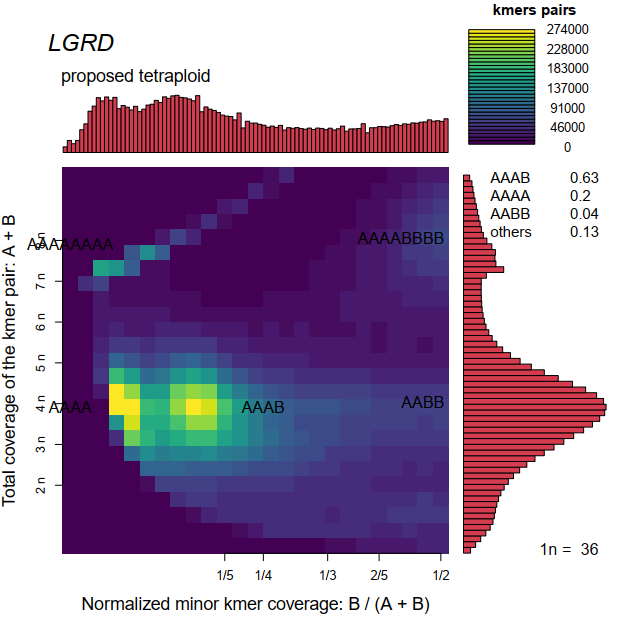 i read the smudgeplot_warnings.txt, get information about provide the information for imporve the software. so...
@chapmanb hi, i running manta , get output file candidateSV.vcf.gz. finding the the genotype of all SV is missing, can you tell me why?
@brentp hi , it report a error when i running smoove. the command is:smoove call -x --name CXL --fasta ../Ref/CXL-1.build.chr.fa --processes 10 --genotype CXL.sort.fixmate.rmdup.bam。 the error is :  Is...
hi @all I have a VCF with three populations. I use sg.pbs to calculate pbs. I got the pbs values, but I want to get CHROM BIN_START BIN_END PBS four...
@sa501428 hi. I want to get the matrix from the hic file, arrange and orient the contig (including forward and reverse) based on the order defined by myself to get...
### bug reports hi guys : Hi all, when I run gatk (version: 4.5.0.0) CombineGVCFs to combine 240 8 ploidy samples gvcf, it reports the error as below  how...
hi everyone: I use render_site to generate my html website but I want to include all dependencies in the html, can you tell me how to achieve it? waiting for...
@philippesanio  The error message shows that line 37 of outputWriter.py, the tbi index cannot be built because the genome is too large. ``` if "gz" in self.output_bed: pysam.tabix_compress(filename_in=tmp_out, filename_out=self.output_bed,...
when I use gatk to identify variants in a bam file converted from gam using vg surject, I get error

@jmarshall when i use [(vg surject)](https://github.com/vgteam/vg) convert gam to bam, then use gatk detect variants, it report error as bleow  the genome no chromosome is longer than 512M,i can't...