methylKit
 methylKit copied to clipboard
methylKit copied to clipboard
R package for DNA methylation analysis

Based on the new badge showing all package deps https://bioconductor.org/packages/release/bioc/html/methylKit.html#since introduced on Bioc we are having 87 deps in total. 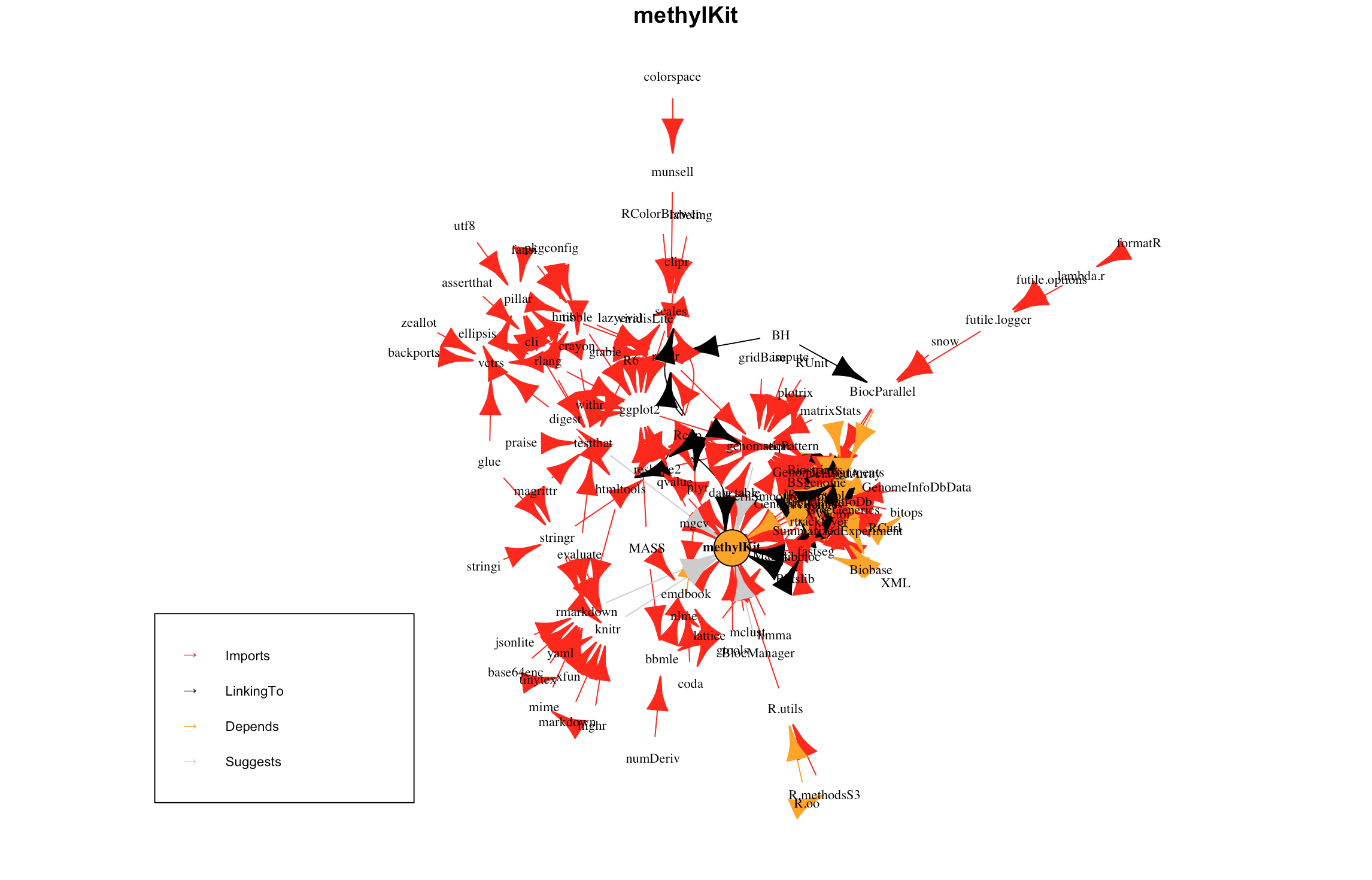 I was dazzled to see that we have ggplot2...
https://github.com/al2na/methylKit/blob/a22fb817a1c244ccba0491b9289064731c801a30/R/bedgraph.R#L78
I have a `methRead` object that is further merged using `unite`. It consists of 5 samples. When running `clusterSamples`, I get the following error: > clusterSamples(Unite.object, dist="correlation", method="ward.D", plot=TRUE,) Error...
See here, the error is not very informative https://groups.google.com/d/msg/methylkit_discussion/9nPkCbPyKLI/a9xCm3mdAwAJ
example: ``` library(methylKit) file.list=system.file("extdata", "test1.myCpG.txt", package = "methylKit") myobj=methRead( file.list, sample.id="test1", assembly="hg18",treatment=c(1,1,0,0), dbtype="tabix") readTabixHeader(getDBPath(myobj)[[1]]) > $dbtype [1] "tabix" $sample.ids [1] "test1" $assembly [1] "hg18" $context [1] "CpG" $resolution [1] "base"...
Right now I saw that it used 2x the size of the input file in memory. This is fine for 40 million CpG sites, but can be quite large for...
Using Methread with mincov=5 results in very odd output - many sites are dropped when compared with mincov=10, whereas the expectation was that mincov=5 would be a superset of mincov=10....
There is this nice package iotools (https://cran.r-project.org/web/packages/iotools/iotools.pdf) which allows fast chunkwise reading and writing from files. It supports connections, i.e. can read and write from /to gzipped files directly. It...
What do you think about adding a method to calculateDiffDSS to use as an input methylBaseDB and to calculate DMCs per chromosome/chunk? if you are ok with it I can...
One cause of that problem could be the call to mclapply when merging result of logreg, because it has to copy the chunks multiple times