tombo
 tombo copied to clipboard
tombo copied to clipboard
"detect_modifications model_sample_compare" output stats file is empty
Hello,
I've run many model_sample_compare runs without problems, but for this run keeps outputting empty stats files, both per read stats files and regular stats files.
Packages in my environment as follow: ont-tombo 1.5.1 py36r36hb3f55d8_0 bioconda python 3.6.10 h0371630_0 h5py 2.9.0 nompi_py36hcafd542_1103 conda-forge
The command line is as follows: tombo detect_modifications model_sample_compare --fast5-basedirs / --statistics-file-basename / --control-fast5-basedirs / --sample-only-estimates / --fishers-method-context 3 / --per-read-statistics-basename /
Below is the job log. [12:31:01] Parsing Tombo index file(s). [12:31:10] Performing two-sample comparison significance testing. [12:31:10] Parsing Tombo index file(s). [12:31:11] Calculating read coverage regions. [12:31:11] Calculating read coverage. [12:31:13] Calculating read coverage. [12:31:13] Performing modified base detection across genomic regions. 0%| | 0/1 [6:22:41<?, ?it/s]
Usually the percentage of process goes up to 100%, but this time it didn't. However, it looked like finished and the time when the output stat files changed is 18:53, which 6hr 22min later from the start time. I got very small sized output stats files and when I opened them with HDF view, they have some directories but empty. I run three times, and the all three runs gave me these results....
Any ideas why this happened?
Thanks, Gabby
There are a number of possibilities here. What was the output of the resquiggle commands for these runs? The main other thing to check would be that these two samples have overlapping reads at sufficient depth. The depth text_output file should help identify any issues here.
Hello marcus,
The commands and the output of the resquiggle for these runs as follows:
tombo resquiggle /single_fast5_dir/ /ref_fasta_file/ --processes 20 --q-score 7 --num-most-common-errors 5 --overwrite --ignore-read-locks
[23:07:52] Loading minimap2 reference.
[23:07:52] Getting file list.
[23:07:56] Loading default canonical ***** RNA ***** model.
[23:07:56] Re-squiggling reads (raw signal to genomic sequence alignment).
5 most common unsuccessful read types (approx. %):
6.8% ( 71051 reads) : Alignment not produced
5.8% ( 60395 reads) : Poor raw to expected signal matching (revert with tombo filter clear_filters)
0.5% ( 4722 reads) : Read event to sequence alignment extends beyond bandwidth
-----
-----
100%|██████████████████████████████████████████████████████████████████████████████████████████████| 1049154/1049154 [11:29:31<00:00, 25.36it/s]
[10:37:28] Final unsuccessful reads summary (13.0% reads unsuccessfully processed; 136168 total reads):
6.8% ( 71051 reads) : Alignment not produced
5.8% ( 60395 reads) : Poor raw to expected signal matching (revert with tombo filter clear_filters)
0.5% ( 4722 reads) : Read event to sequence alignment extends beyond bandwidth
[10:37:28] Saving Tombo reads index to file.
And this is the visualized coverage text_output file using IGV. The upper track is 'coverage.control.plus', lower track is 'coverage.sample.plus.
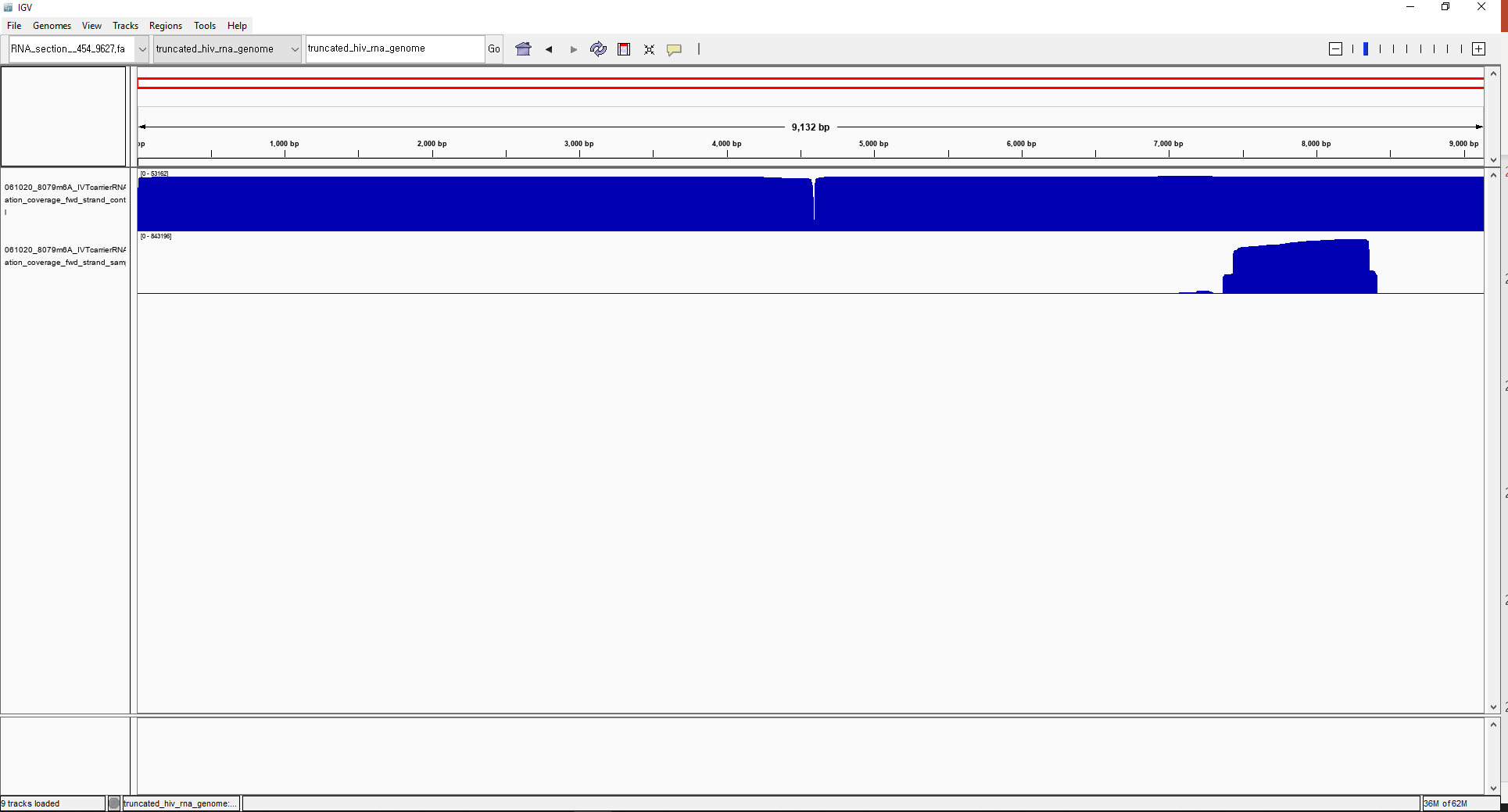
I tried another file types other than 'coverage' with text_output browser_files, but the output files don't have any data.
Thanks, Gabby
This is indeed quite odd. I'm wondering if there might be some silent failure mode here. 800,000X coverage is likely far above the coverage you need for sufficient results. I wonder if you might downsample these runs to a lower value (see tombo filter level_coverage command docs here; maybe ~10,000X coverage) to see if the command completes successfully. I would recommend saving the hidden tombo index file to get the original results back with default filters if required as described here.
If this works then it might be worth looking at where this might fail. I would've guessed that this level of coverage might have thrown a memory error. Is there any chance that this was run on a job queuing system (SGE/UGE/slurm) and may have been silently killed by the job scheduler?
After downsampling, I got stats file and per read stats file properly. When the control coverage is higher than sample coverage, model_sample_compare method runs without error.
Thank you so much! Gabby
It shouldn’t matter which sample has more coverage. My suspicion is that this is a memory error that python couldn’t catch.