bedtools2

bedtools2 copied to clipboard
bamtofastq creates duplicate reads
Hello,
I found an issue regarding bamtofastq in bedtools (v2.30.0). When I convert a bam file into a fastq file, it seems to create duplicate reads. You can see that there are duplicate read in the fastq from bedtools. The file size of the fastq from bedtools seems double also.
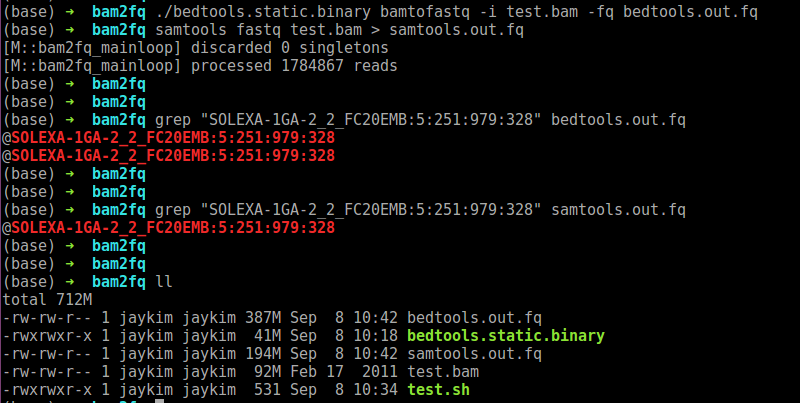
I am attaching a shell script for the reproducibility. The following is the version of tools I use.
bedtools - v2.30.0 samtools - 1.16 htslib - 1.16
set -eou pipefail
# download some bam file from uscs
wget -O test.bam http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeUwRepliSeq/wgEncodeUwRepliSeqBg02esG1bAlnRep1.bam
# bam2fq
./bedtools.static.binary bamtofastq -i test.bam -fq bedtools.out.fq
samtools fastq test.bam > samtools.out.fq
# grep count
echo "this is from the bedtools output"
grep "SOLEXA-1GA-2_2_FC20EMB:5:251:979:328" bedtools.out.fq
echo ""
echo "this is from the samtools output"
grep "SOLEXA-1GA-2_2_FC20EMB:5:251:979:328" samtools.out.fq
Hello bedtools devs,
I also see duplicate reads using v2.30.0. I do not see duplicates using v2.26.0.
I should note that I have an unaligned BAM file, so the issue is not related to the number of mappings.