fastp
 fastp copied to clipboard
fastp copied to clipboard
Duplication rate differs up to 30% from that of Fastqc for single end reads
I found a quite big difference in the duplication between Fastp and Fastqc. For all my ~40 SE RNAseq samples, the rate is around 10-30% lower in Fastp compared to Fastqc. Is there an explanation for this or is it a bug?

1, duplication evaluation for SE data is always inaccurate, since you never know where is the another end of the DNA fragment. For RNA data, it is more serious.
2, I found FastQC used trimmed data, the trimming will increase duplication level since it make it shorter. You can compare the original FASTQ.
@sfchen
Fastqc (Trimmed) refers to the output of Fastp which I ran only with default parameters. Sorry for the confusion.
Has anyone else seen the same discrepancy between Fastqc and Fastp?
@sfchen
Multiqc says that the Fastp duplication rate was calculated on the "filtered" reads which I understand as the reads outputted by Fastp. Am I understanding something wrong or is this misleading description in Multiqc?

I also checked the Fastqc duplication rate in the original (raw) data and it corresponds almost exactly with the Fastqc duplication rate after Fastp processing.
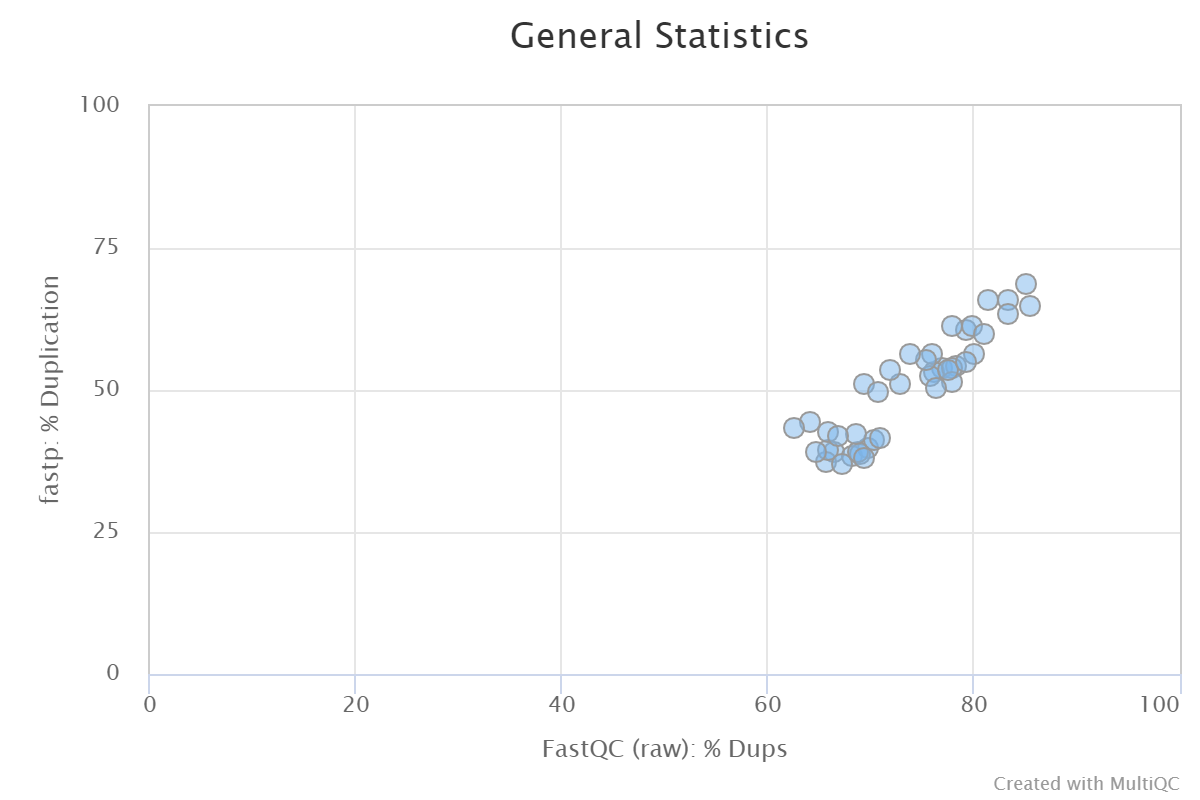
I think you missed this: I also checked the Fastqc duplication rate in the original (raw) data and it corresponds almost exactly with the Fastqc duplication rate after Fastp processing.
Hi, I am using this issue as I am facing a similar problem. For SE reads the duplication rates reported by fastp and FastQC (on the same raw reads) differ significantly:
fastp --in1 test.fq.gz --out1 test_fastp.fq.gz
Detecting adapter sequence for read1...
No adapter detected for read1
Read1 before filtering:
total reads: 20271682
total bases: 3040752300
Q20 bases: 3040752300(100%)
Q30 bases: 3040752300(100%)
Read1 after filtering:
total reads: 20271682
total bases: 3040752300
Q20 bases: 3040752300(100%)
Q30 bases: 3040752300(100%)
Filtering result:
reads passed filter: 20271682
reads failed due to low quality: 0
reads failed due to too many N: 0
reads failed due to too short: 0
reads with adapter trimmed: 0
bases trimmed due to adapters: 0
Duplication rate (may be overestimated since this is SE data): 3.83467%
JSON report: fastp.json
HTML report: fastp.html
fastp --in1 test.fq.gz --out1 test_fastp.fq.gz
fastp v0.23.1, time used: 114 seconds
Running FastQC on the same input file I get the following result:
fastqc test.fq.gz -o qc/

Any ideas?
maybe try this computation mentioned here: https://www.biostars.org/p/83842/