varianttools
 varianttools copied to clipboard
varianttools copied to clipboard
Error in simulation in model resample
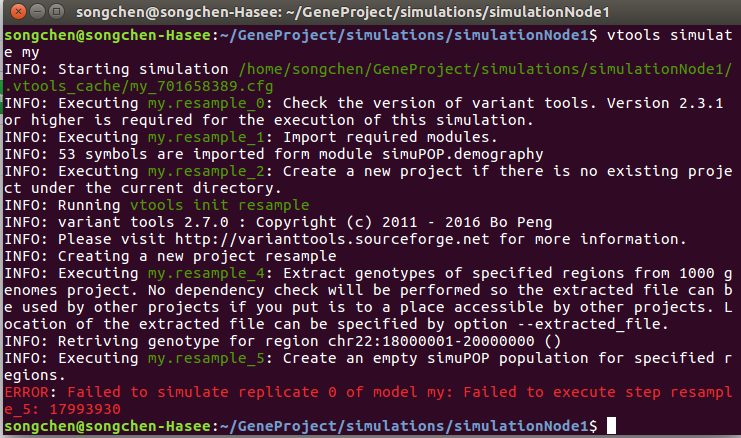
Hello, variant tool users! I wrote a pipeline which is basically the same as Peng2014_ex1.resample. The only difference is that I adjusted some parameters. When I run the pipeline, it shows the follow error: ERROR: Failed to simulate replicate 0 of model my: Failed to execute step resample_5: 17993930 Here is the terminal:
Here is the pipeline I used: my.pipeline
[pipeline description]
description=A pipleline made by Songchen, whose goal is to mimic the Peng_2014_ex1
pipeline but change it so that this pipeline can be used for generating DNA sequence
which can be used for exploiting compressing algorithm for huge sample number DNA data.
resample_description=A simulation model that extracts genotypes within the
sepecified regions from the 1000 genomes project, and expands it very
rapidly to mimick a resampling-based simulation.
[DEFAULT]
regions=chr22:18000001-20000000
regions_comment=One or more chromosome regions (separated by ',') in the format of
chr:start-end (e.g. chr21:33031597-33041570), or Field:Value from a region-based
annotation database (e.g. refGene.name2:TRIM2 or refGene_exon.name:NM_000947).
Please visit http://varianttools.sourceforge.net/Simulation for detailed
description of this parameter.
scale=13
scale_comment=Scaling factor to speed up the simulation by scaling down the
simulation while boosting mutation, selection and recombination rates.
extracted_vcf=extracted.vcf
extracted_vcf_comment=Filename (with dir) to save genotypes (.vcf file extracted by tabix
command) for the resample model. This file will be automatically created and
reused if it already exists. You will need to remove this file if you run the
same pipeline using another region.
[*_0]
action=CheckVariantToolsVersion('2.3.1')
comment=Check the version of variant tools. Version 2.3.1 or higher is required
for the execution of this simulation.
[*_1]
action=ImportModules('simuPOP.demography')
comment=Import required modules.
[resample_2]
input_emitter=EmitInput(select=${:not glob.glob('*.proj')})
action=RunCommand('vtools init resample')
comment=Create a new project if there is no existing project under the current
directory.
[resample_4]
action=ExtractVCF(
sourceURL='gene1000chr22.vcf.gz',
regions='%(regions)s',
output='%(extracted_vcf)s')
input_emitter=EmitInput(select=${:not os.path.isfile('%(extracted_vcf)s')})
output='%(extracted_vcf)s'
comment=Extract genotypes of specified regions from 1000 genomes project. No
dependency check will be performed so the extracted file can be used by
other projects if you put is to a place accessible by other projects. Location
of the extracted file can be specified by option --extracted_file.
[resample_5]
action=CreatePopulation( importGenotypeFrom='%(extracted_vcf)s',
regions='%(regions)s',
output='cache/my_resample_init_${seed}.pop')
output='cache/my_resample_init_${seed}.pop'
comment=Create an empty simuPOP population for specified regions.
[resample_6]
action=EvolvePopulation(
output='my_resample_sampled_${seed}.pop',
mutator=RefGenomeMutator(regions='%(regions)s', model='JC69', rate=[1.8e-8 * %(scale)s * 4. / 3.]),
selector=ProteinSelector('%(regions)s', s_missense=0.0001 * %(scale)s,
s_stoploss=0.0001 * %(scale)s, s_stopgain=0.001 * %(scale)s),
transmitter=FineScaleRecombinator(regions='%(regions)s', scale=%(scale)s),
demoModel=ExponentialGrowthModel(T=4,
NT=10000)
)
comment=Expand the population exponentially to d a large population
in 10 generations. Mutations and recombinations are allowed and
a selection model that only select against stopgain mutations are
used.
[resample_7]
action=ExportPopulation(
output='newSimulatedPopulation.vcf'
)
[resample_999]
action=RemoveIntermediateFiles(['my_resample_sampled_${seed}.pop'])
comment=Remove intermediate populations to save diskspace.
The sourceURL file I used gene1000chr22.vcf.gz is downloaded from the 1000 genome project. http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/ the 22nd chromosome.
To locate the error, I checked the extracted .vcf file, 'extracted.vcf'. I exported all the positions in that file, and even though in the pipeline I set the region as chr22:18000001-20000000, it contains a 17993930 in 'extracted.vcf'. I think this is the reason for that error. Also I checked the definition of the pipeline action 'ExtractVCF' and found out that it is the 'tabixFetch' funtion who extracts the vcf file. So maybe there is something wrong with the tabixFetch function and the variant tools?
Thanks in advance for any help!!!
Are you using python 3? If so, are you using the latest version from github? We recently fixed a python3 related issue (https://github.com/vatlab/VariantTools/issues/61).
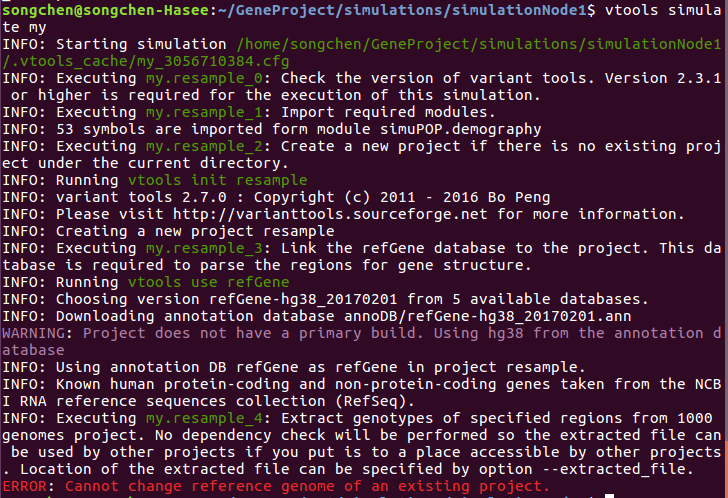
Update, I didn't use the refGene database before, so I tried with adding back the action
[resample_3] action=RunCommand('vtools use refGene') comment=Link the refGene database to the project. This database is required to parse the regions for gene structure.
This time, it reports the error 'ERROR: Cannot change reference genome of an existing project.'
@BoPeng Yes, I think I used the newest version. And I am the person who raised that issue. And I'm using python 3. Thank you for the fast response.
The reason for the error message is that VST assumes hg19 because the variants will be resampled (evolved) from a thousand genomes data which is based on hg19. Your version of refGene-hg38 then not work. You will have to either use hg19 version of refGene (add version number to vtools use) or find hg38 version of 1000Genomes.
@BoPeng I am using the dataset 'ALL.chr22.phase3_shapeit2_mvncall_integrated_v5a.20130502.genotypes.vcf.gz' so I guess the annotation 'thousandGenomes-hg19_20130502.DB.gz' in http://bioinformatics.mdanderson.org/Software/VariantTools/repository/annoDB/ is the one I should use?
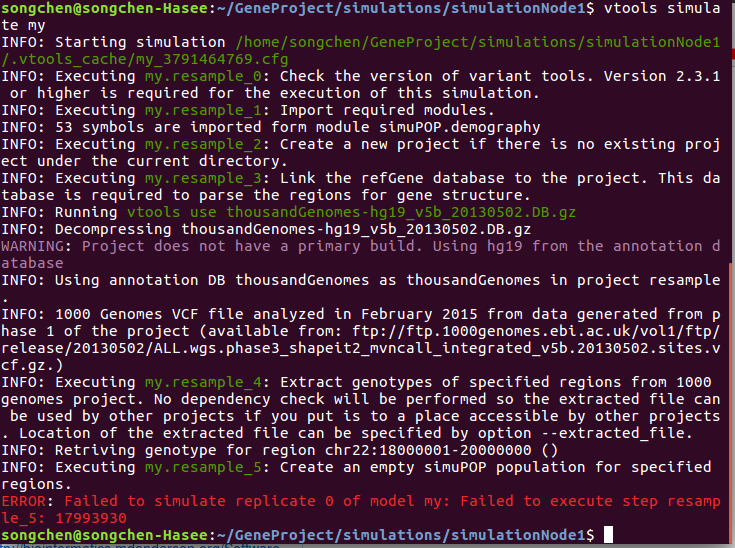
@BoPeng Thank you for the instructions. I am using the thousandGenomes-hg19_v5b_20130502 annotation now and it works, but I still got the error regarding extracting the right region.
@BoPeng The directory I run VTS contains the following files:
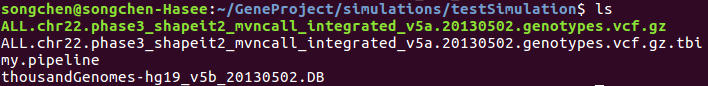
the two gene data file were downloaded from http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/ The DB file is downloaded from http://bioinformatics.mdanderson.org/Software/VariantTools/repository/annoDB/ in gzziped format and has already been decompressed. The pipeline my.pipeline is as follow:
[pipeline description]
description=A pipleline made by Songchen, whose goal is to mimic the Peng_2014_ex1
pipeline but change it so that this pipeline can be used for generating DNA sequence
which can be used for exploiting compressing algorithm for huge sample number DNA data.
resample_description=A simulation model that extracts genotypes within the
sepecified regions from the 1000 genomes project, and expands it very
rapidly to mimick a resampling-based simulation.
[DEFAULT]
regions=chr22:18000001-20000000
regions_comment=One or more chromosome regions (separated by ',') in the format of
chr:start-end (e.g. chr21:33031597-33041570), or Field:Value from a region-based
annotation database (e.g. refGene.name2:TRIM2 or refGene_exon.name:NM_000947).
Please visit http://varianttools.sourceforge.net/Simulation for detailed
description of this parameter.
scale=13
scale_comment=Scaling factor to speed up the simulation by scaling down the
simulation while boosting mutation, selection and recombination rates.
extracted_vcf=extracted.vcf
extracted_vcf_comment=Filename (with dir) to save genotypes (.vcf file extracted by tabix
command) for the resample model. This file will be automatically created and
reused if it already exists. You will need to remove this file if you run the
same pipeline using another region.
[*_0]
action=CheckVariantToolsVersion('2.3.1')
comment=Check the version of variant tools. Version 2.3.1 or higher is required
for the execution of this simulation.
[*_1]
action=ImportModules('simuPOP.demography')
comment=Import required modules.
[resample_2]
input_emitter=EmitInput(select=${:not glob.glob('*.proj')})
action=RunCommand('vtools init resample')
comment=Create a new project if there is no existing project under the current
directory.
[resample_3]
action=RunCommand('vtools use thousandGenomes-hg19_v5b_20130502.DB')
comment=Link the refGene database to the project. This database is required
to parse the regions for gene structure.
[resample_4]
action=ExtractVCF(
sourceURL='ALL.chr22.phase3_shapeit2_mvncall_integrated_v5a.20130502.genotypes.vcf.gz',
regions='%(regions)s',
output='%(extracted_vcf)s')
input_emitter=EmitInput(select=${:not os.path.isfile('%(extracted_vcf)s')})
output='%(extracted_vcf)s'
comment=Extract genotypes of specified regions from 1000 genomes project. No
dependency check will be performed so the extracted file can be used by
other projects if you put is to a place accessible by other projects. Location
of the extracted file can be specified by option --extracted_file.
[resample_5]
action=CreatePopulation(
importGenotypeFrom='%(extracted_vcf)s',
regions='%(regions)s',
output='cache/my_resample_init_${seed}.pop')
output='cache/my_resample_init_${seed}.pop'
comment=Create an empty simuPOP population for specified regions.
[resample_6]
action=EvolvePopulation(
output='my_resample_sampled_${seed}.pop',
mutator=RefGenomeMutator(regions='%(regions)s', model='JC69', rate=[1.8e-8 * %(scale)s * 4. / 3.]),
selector=ProteinSelector('%(regions)s', s_missense=0.0001 * %(scale)s,
s_stoploss=0.0001 * %(scale)s, s_stopgain=0.001 * %(scale)s),
transmitter=FineScaleRecombinator(regions='%(regions)s', scale=%(scale)s),
demoModel=ExponentialGrowthModel(T=4,
NT=10000)
)
comment=Expand the population exponentially to d a large population
in 10 generations. Mutations and recombinations are allowed and
a selection model that only select against stopgain mutations are
used.
[resample_7]
action=ExportPopulation(
output='newSimulatedPopulation.vcf'
)
[resample_999]
action=RemoveIntermediateFiles(['my_resample_sampled_${seed}.pop'])
comment=Remove intermediate populations to save diskspace.
The command line I use is just vtools simulate my and the error looks like below:
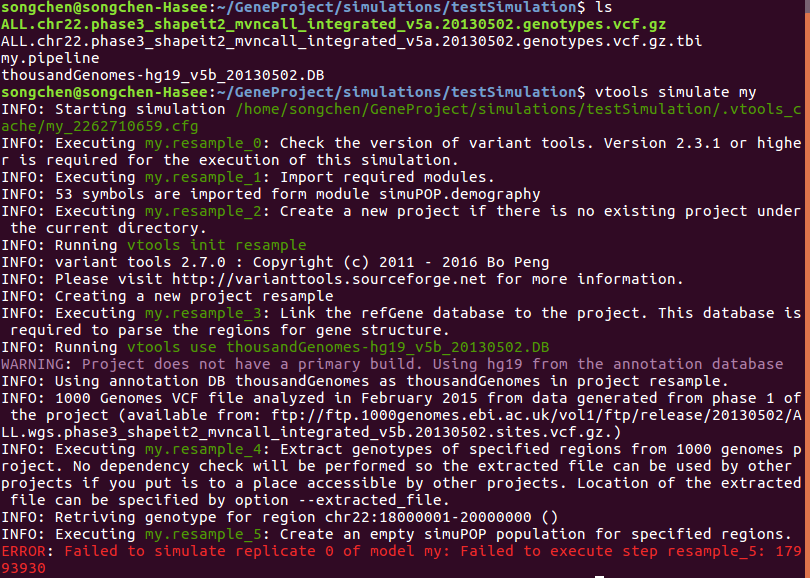
Variant_tools is installed from the github code, and simupop version is 1.1.8.3.
After running simulation, the directory contains the following files:
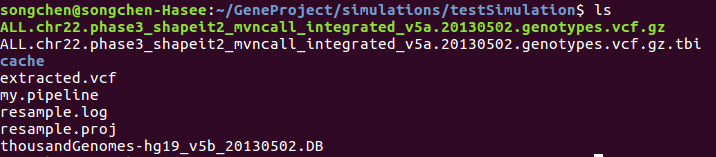
To see which positions are in 'extracted.vcf' I used bcftools query -f '%POS\n' extracted.vcf > positions, then in the outputted positions file, I saw:

position 17993930 is not in the region I set in my.pipeline, but somehow it is extracted.
Thanks for any advice!
Finally get some time to work on this and I can now reproduce the problem. I think this is a bug with tabix that has been observed by others.
I do not get it.
tabix ALL.chr22.phase3_shapeit2_mvncall_integrated_v5a.20130502.genotypes.vcf.gz 22:18000000-20000000
outputs the following as the first line
22 17993930 esv3647221 T <CN0> 100 PASS AC=1;AF=0.000199681;AN=5008;CIEND=-150,150;CIPOS=-150,150;CS=DEL_union;END=18000439;NS=2504;SVTYPE=DEL;DP=13526;EAS_AF=0;AMR_AF=0;AFR_AF=0.0008;EUR_AF=0;SAS_AF=0;VT=SV GT 0|0 0|0 0|0 0|0
0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0
0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0 0|0
so it seems that there is something wrong with tabix itself?
OK, I have pushed a patch to ignore loci that are extracted by tabix that are outside of the specified region. The workflow should work now.