tripal
 tripal copied to clipboard
tripal copied to clipboard
1245 tv3 newick loader
Bug Fix
Issue #1245
Description
This pull request contains a number of bug fixes and enhancements to the Newick phylogenetic tree loader. This started as an attempt to get taxonomy trees to link to organisms, which had not been implemented for Newick loaded trees, but a number of issues showed up which needed fixing.
- Fuller implementation of job logging to allow output to go to the "jobs" page and not just to drush output.
- Additional log messages to show if leaf nodes were or were not associated with features or organisms.
- Taxonomy trees loaded from Newick files now link to organisms. A new function does the name to organism lookup
chado_phylogeny_lookup_organism_by_name()which in turn callschado_unabbreviate_infraspecific_rank()from pull #1244 - Fixed the bug in the regular expression option.
- Various typo fixes.
- The "Feature Name Regular Expression" has been expanded to also work on taxonomy trees loaded from Newick files.
- The
operation__phylotree_vis_formatterfield now saysClick a species to view its species page.only for taxonomy trees, it now displaysClick a node label to view its page.for all other types of phylogenetic trees. - There is the existing option to load the tree with the loader job, or as a separate job. I flipped the logic so that unchecked is the default, and the default is to load everything at once. This option is not currently available to the drush implementation, so the change should not break anything. (
load_nowbecomesload_later). - Some of the error messages have been enhanced to show the value that caused the error.
- There's a thing added to
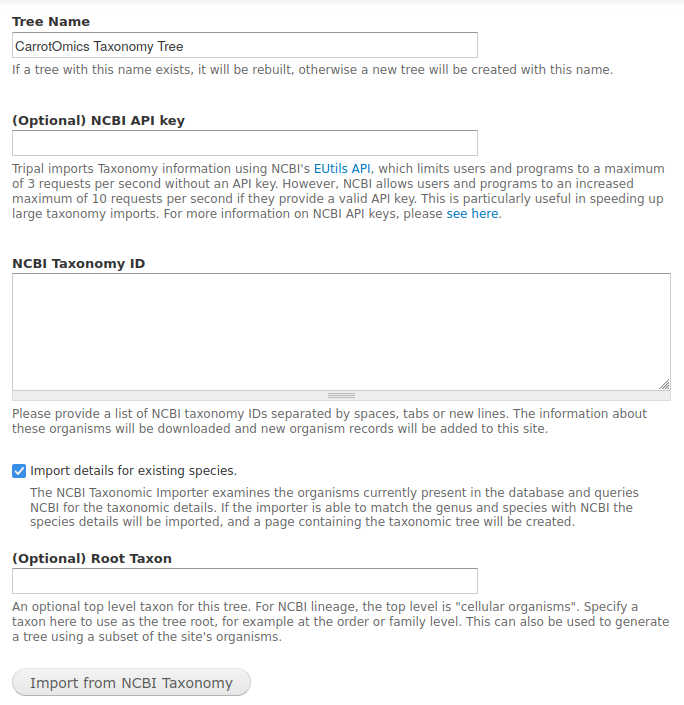
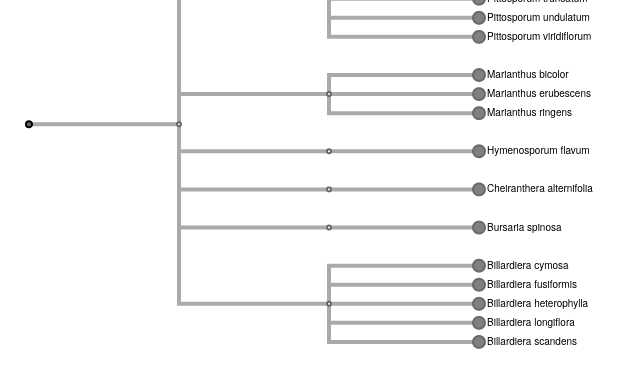
tripal.moduleto make sure the legacy'file'value is present before adding it. - NCBI Taxonomy importer form adds options for tree name and root taxon. This allows a site to have a subset of the organisms in a tree, such as a separate tree for each family. I added an example image below for the new input form, and for a tree with the family as the root. The default for the tree name is unchanged from before for backwards compatibility, although really there should be a space added.
- Queries to NCBI are now wrapped in a retry loop, to handle rare cases when an internet query goes bad. This is quite rare for me, but does happen.
- On rare occasions a query to NCBI has no full match, but NCBI returns a partial match which yields an incorrect taxid. This is now checked for, and the invalid taxid discarded. Example, this returns a taxid for a bacterium: https://www.ncbi.nlm.nih.gov/entrez/eutils/esearch.fcgi?db=taxonomy&term=Torilis+arvensis+subspecies+neglecta
- Some conflicting comments as to whether an organism tree uses 'taxonomy' or 'organism' when loading has been changed to always use 'taxonomy'. Also this setting is now only passed through
$options['leaf_type']consistently throughout. - Implemented the "to-do" of using stored NCBI taxid for site organisms, when present. Though this then can lead to a difference in site vs. NCBI taxon names. So...
- In the case of heterotypic or homotypic synonyms, the taxon name stored in chado is used in preference to that returned from the NCBI taxid query, in order to keep the site consistent.
- Phylotree leaf nodes can now link to stock in addition to feature and organism. Useful for loading newick tree files linked to germplasm accessions.
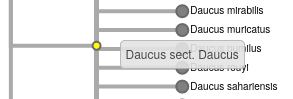
- If internal tree nodes have labels, they will display with a mouse-over. Example image shown below.
Example images for item #11. Added two input fields for tree name, and root taxon.
Part of an example sub-tree with the root at the family level (Pittosporaceae).
For item 18: Example image showing mouseover of interior node of a taxonomic tree
Item 3 on my list will need to be modified if pull #1328 is accepted, this implements a general api function to look up an organism from its name, and would replace most of the function chado_phylogeny_lookup_organism_by_name() I created in tripal_chado.phylotree.api.inc
This pull request is now dependent on #1328 being merged first, so testing will fail until that happens.
@dsenalik can you provide a sample newick file I can use for testing and a set of instructions to test?
@spficklin I actually created two basic automated tests for this loader, which previously had no tests. (Hopefully this can help with porting to tripal 4 eventually where we need full test coverage). Looks like they passed, yay! The second test checks items 3 and 6 at the top of this pull request, to link directly to organisms and the regex applying to this also. There are a lot of things I did here, and the hardest part of testing or writing a testing protocol is to create the organisms and features before you can test the loader. Hopefully this is a start...