There are differences between the simulation results of Pybamm and those of COMSOL.
Dear PyBamm Support Team, I have recently been learning about PyBamm-based lithium-ion electrochemical DFN simulation modeling, but I have encountered some issues, which are outlined below:
-
When I compared the simulation results of COMSOL and PyBamm using the parameters from "Battery_Management_Systems_Volume_1_Battery_Modeling_Plett_Gl" (Section 4.13, Model Simulations), I noticed a significant discrepancy in the output voltage between the two. However, their output open-circuit voltages (OCV) are very close.
-
To identify the source of this discrepancy, I verified using AMESIM with the same parameters. The results from COMSOL and AMESIM showed good consistency. I suspect this difference may be related to numerical calculation methods. It is known that COMSOL uses the finite element method (FEM), while PyBamm uses the finite volume method (FVM). The theoretical section on DFN in the AMESIM user manual mentions the concept of control volumes, so I hypothesize that AMESIM also uses the finite volume method.
-
As shown in the figures below, I compared two cases: one with an initial state of 80% SOC discharged at 1C for 1000 seconds, and another discharged at 4C until reaching the cut-off voltage of 2.5V. The simulation data has been attached. For the 1C discharge case, the maximum error between COMSOL and PyBamm is 10mV; for the 4C discharge case, the maximum error between the two is 45mV. In contrast, the maximum error between COMSOL and AMESIM is 0.7mV for 1C discharge and 7mV for 4C discharge.
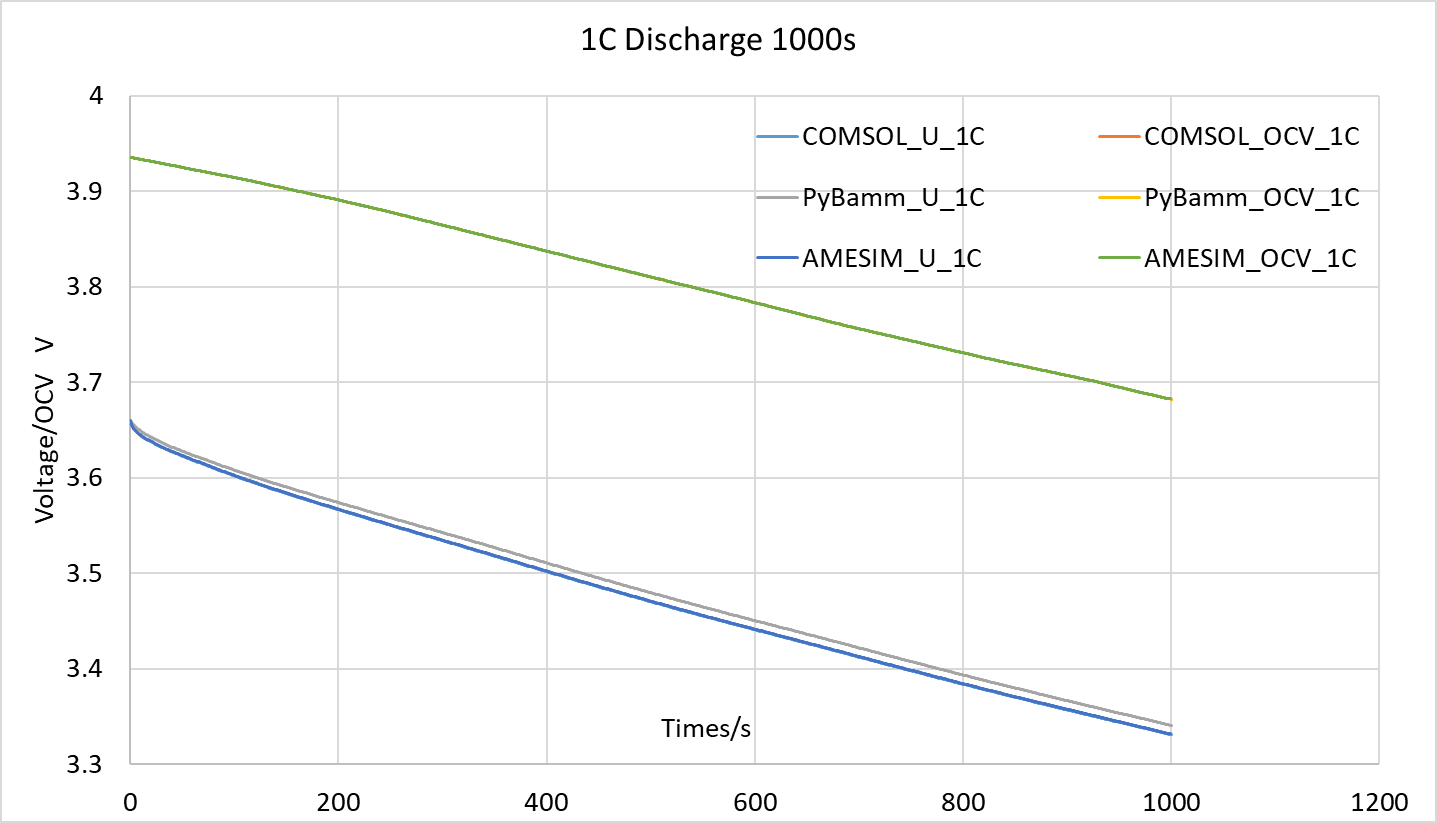
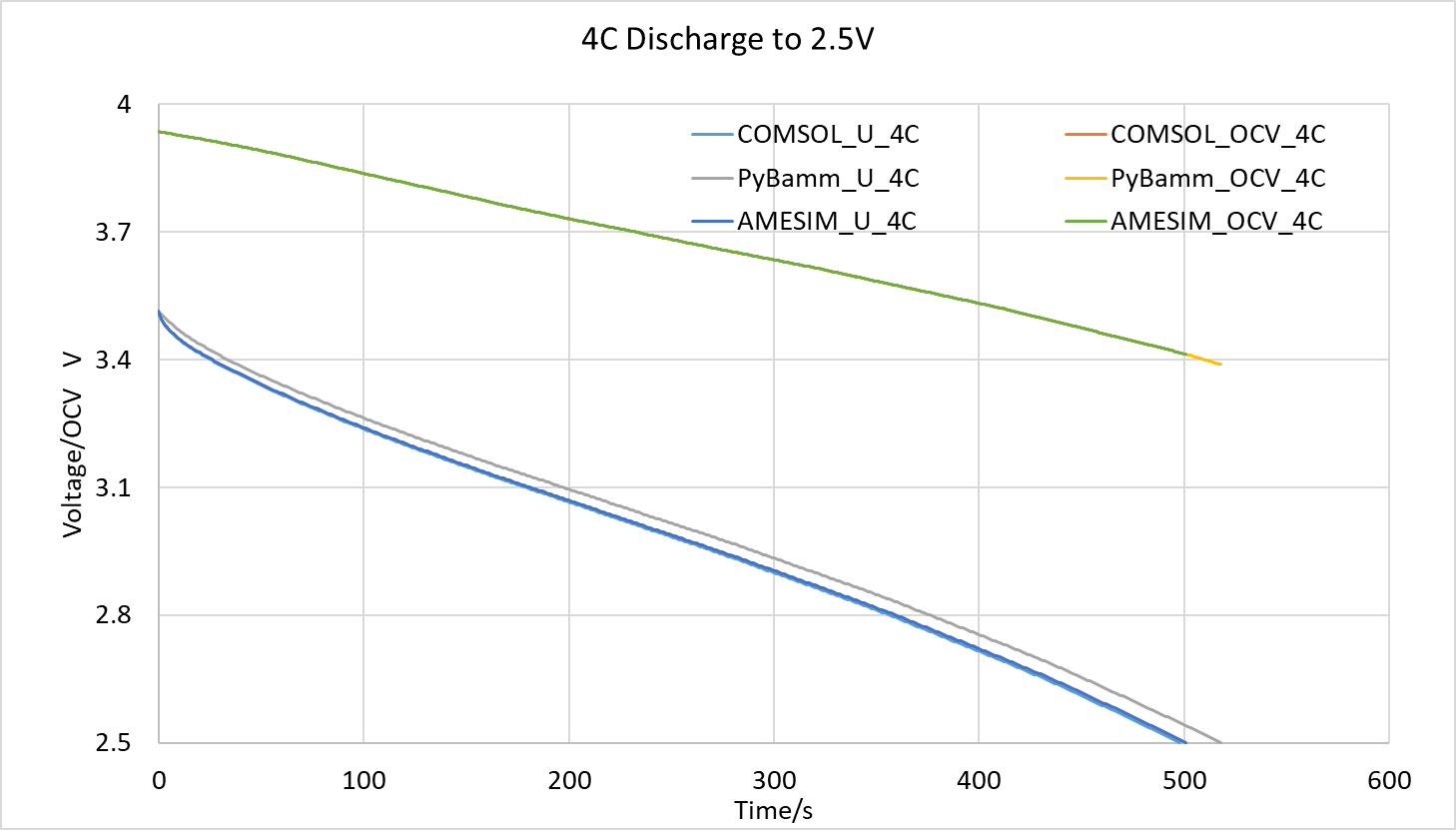
-
I found in the PyBamm examples (https://docs.pybamm.org/en/stable/source/examples/notebooks/models/compare-comsol-discharge-curve.html) that the simulation results of PyBamm and COMSOL are in excellent agreement.
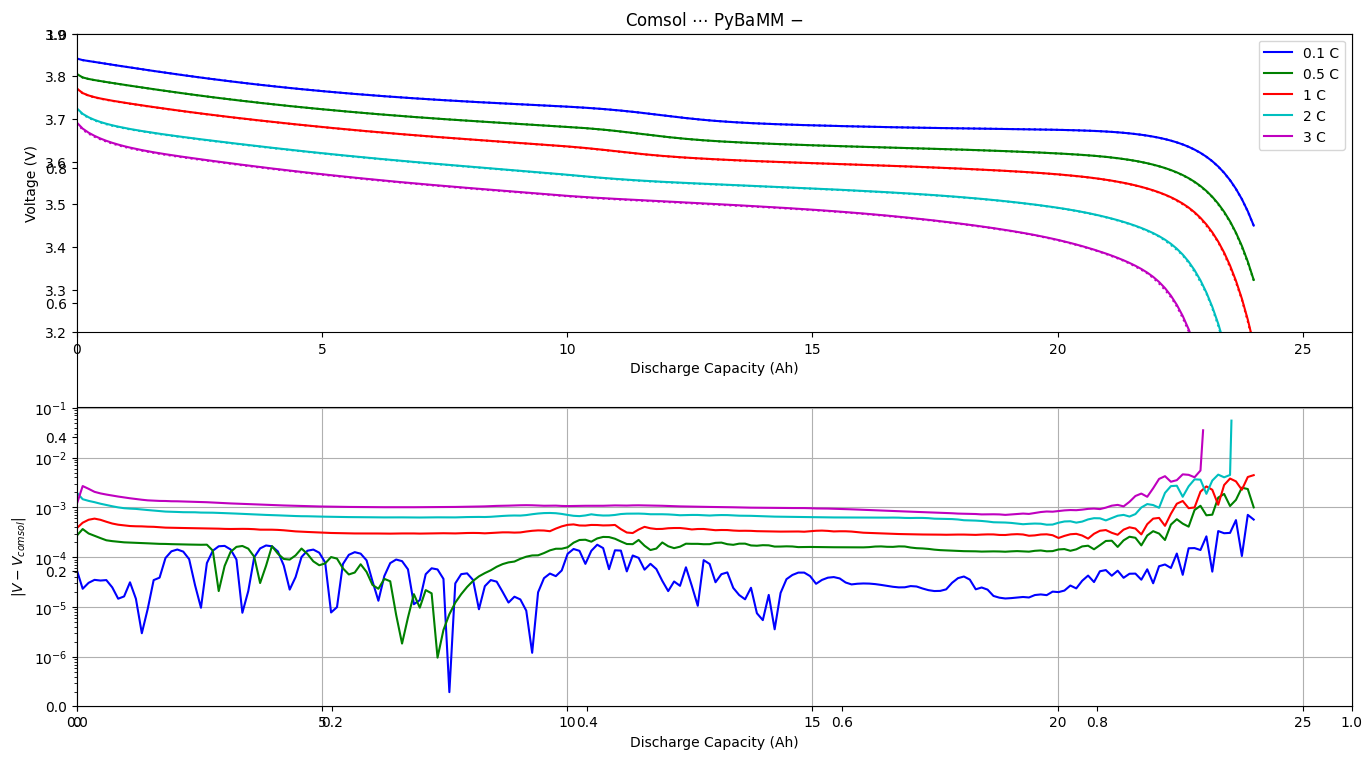
However, I cannot reproduce this consistency. Despite a series of troubleshooting steps, I still haven’t found a solution. Therefore, I am reaching out to the PyBamm support team for advice and would greatly appreciate your assistance.
Verification Model File :https://github.com/Wolframscs/PIC-Vault/blob/main/PyBamm/Pybamm_Verification.zip Note: The .mph model was created in COMSOL 6.3, while the .ame model was created in AMESIM 2404 version.
I have attached my Pybamm parameter file, the COMSOL simulation model file, and the AMESIM simulation model file in the link:https://github.com/Wolframscs/PIC-Vault/blob/main/PyBamm/Pybamm_Verification.zip
If no one answers this question, then it is certain that although Pybamm simulation results can always be made consistent with experimental results through parameter identification, comparisons with simulation results from other commercial software indicate that Pybamm is wrong.
Its very hard to compare with your file and results for me. Would be easier to comment if you plotted Current [A] vs time and had a single file with the scalar parameters and plots of the functional parameters side by side.
You suspect its solver differences? How can we know its not parameter differences, model/options differences without a side by side comparison.
I can't actually open the diff file structures. I am confident you'll get more feedback if you allow us to inspect things in a comparable way......ideally as text or in your excel file
I suspect the differences are due to differences in how parameters are defined. For example, when I did the comparison in the examples, there was a factor of 2 difference in how the kinetics were defined in PyBaMM and COMSOL.
comparisons with simulation results from other commercial software indicate that Pybamm is wrong
I sense your frustration here, but I don't think that "PyBaMM is wrong" is the right conclusion. We have been doing work on the Battery Parameter eXchange project to help tackle this exact issue of comparing tools. BPX is an open standard that provides a common definition of physics-based battery models and their parameters, and the supporting tools allow BPX-compliant parameter sets (a BPX JSON of parameters) to be imported into different simulation tools. As far as I know, COMSOL doesn't yet have a native BPX reader, but taking a look at the standard might help you get to the bottom of things.
After some research, I found that the definition of "Activity factor concentration variation" in Pybamm is different from that in COMSOL. Moreover, in the theoretical formula of Pybamm, there is no additional term for the activation factor. For example, in Battery_Parameter_eXchange_v1.0_July2025, the charge conservation in the electrolyte is:
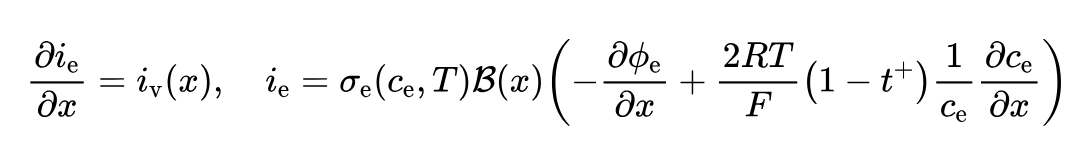
In DFN.ipynb, for: The DFN comprises equations for charge and mass conservation in the solid the solid and electrolyte, and also prescribes behaviour for the electrochemical reactions occurring on the interface between the solid an electrolyte. For more information please see [2] or other standard texts. Below we summarise the model, with all parameters give in the table at the end of this notebook. Here we use a roman subscript $\text{k} \in \text{n, s, p}$ is used to denote the regions negative electrode, separator, and positive electrode, respectively. The model equations for the DFN read:
Charge conservation:
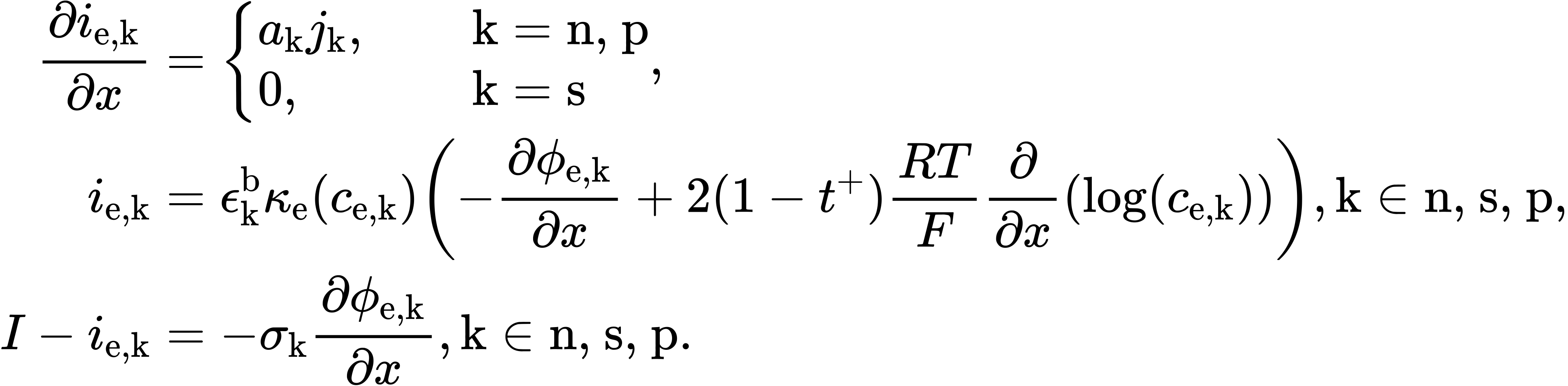
The theoretical formulas provided by PyBamm do not include:
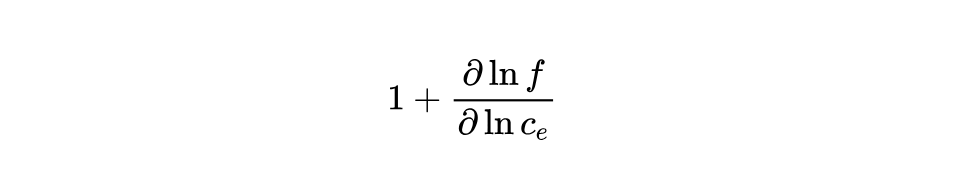
By reviewing the source code, I discovered that the PyBamm code actually defines the "thermodynamic_factor" as follows:
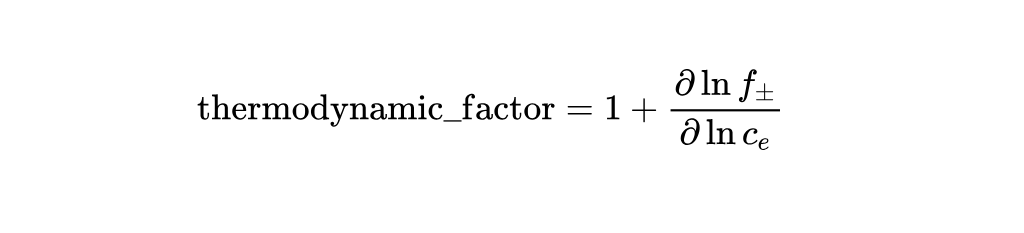 The source code defined in lithium_ion_parameters.py is:
The source code defined in lithium_ion_parameters.py is:
def chi(self, c_e, T):
"""
Thermodynamic factor:
(1-2*t_plus) is for Nernst-Planck,
2*(1-t_plus) for Stefan-Maxwell,
see Bizeray et al (2016) "Resolving a discrepancy ...".
"""
return (2 * (1 - self.t_plus(c_e, T))) * (self.thermodynamic_factor(c_e, T))
def chiRT_over_Fc(self, c_e, T):
"""
chi * (1 + Theta * T) / c,
as it appears in the electrolyte potential equation
"""
tol = pybamm.settings.tolerances["chi__c_e"]
c_e = pybamm.maximum(c_e, tol)
return (self.R * T / self.F) * self.chi(c_e, T) / c_e
def t_plus(self, c_e, T):
"""Cation transference number"""
inputs = {"Electrolyte concentration [mol.m-3]": c_e, "Temperature [K]": T}
return pybamm.FunctionParameter("Cation transference number", inputs)
def thermodynamic_factor(self, c_e, T):
"""Thermodynamic factor"""
inputs = {"Electrolyte concentration [mol.m-3]": c_e, "Temperature [K]": T}
return pybamm.FunctionParameter("Thermodynamic factor", inputs)
When implemented in COMSOL, it takes the form of:
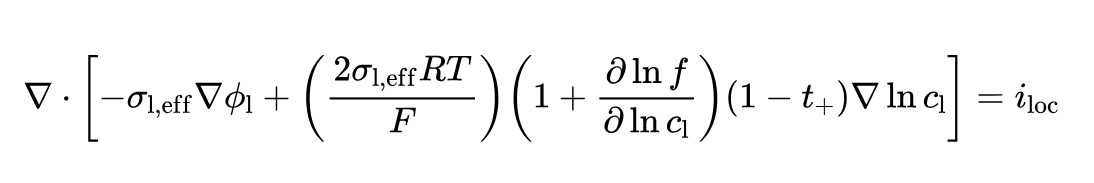 Therefore, the way of defining "Activity factor concentration variation" in COMSOL and Pybamm is different.
Therefore, the way of defining "Activity factor concentration variation" in COMSOL and Pybamm is different.
When defining the Activity factor concentration variation in the PyBamm parameters, the +1 should be included in the defined thermodynamic_factor: If in COMSOL dlnfdlnc = 1, then in PyBamm, the Thermodynamic factor should be defined as 1 + 1 = 2, for example:
"Thermodynamic factor":dlnfdlnc+1,
Through these adjustments, I found that the two simulation results have improved even further:
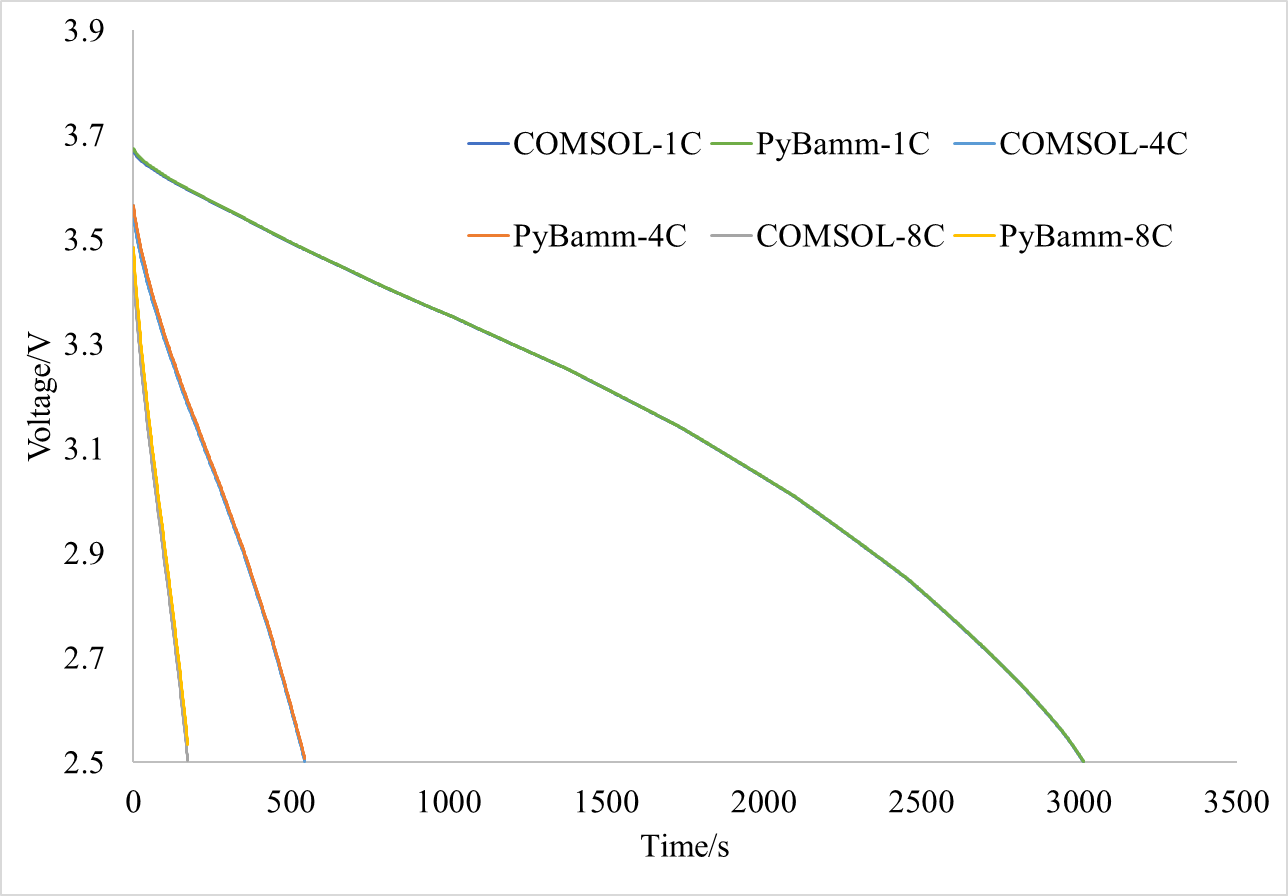
However, at high magnification, there is still a considerable difference between the two: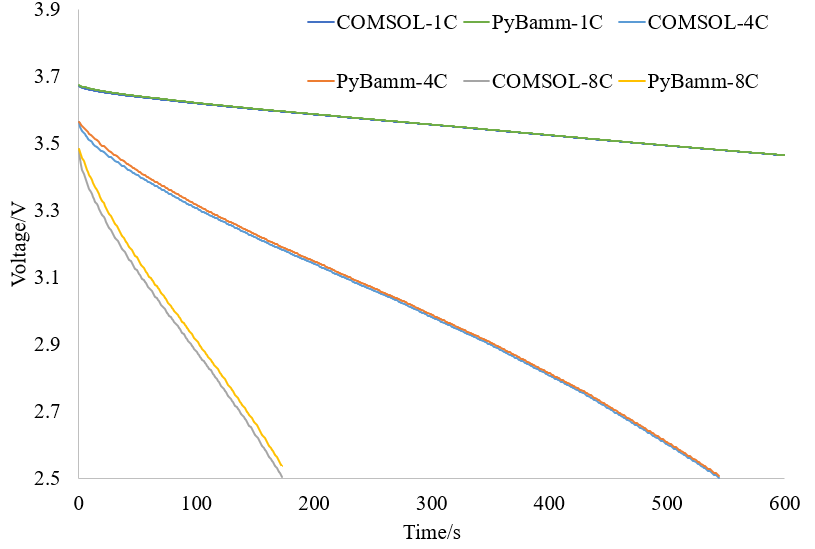 I will further debug and investigate the cause. Although it has been stated in the CHANGELOG:
I will further debug and investigate the cause. Although it has been stated in the CHANGELOG:
- Renamed parameter "1 + dlnf/dlnc" to "Thermodynamic factor". (#2727)
However, I believe that the current theoretical formula provided by PyBamm does not include the variation of Activity factor concentration, and this remains a problem. The complete charge conservation in the electrolyte should be:
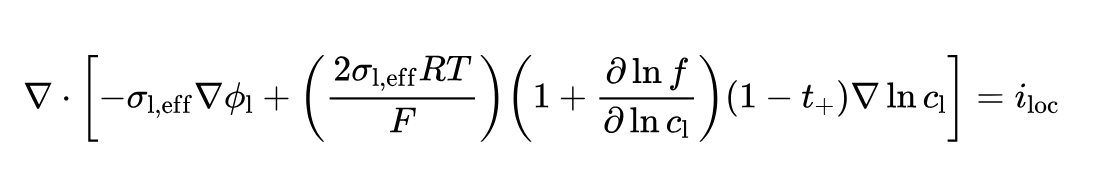
@rtimms @mpegis