New Hybrid Modules
This builds upon ClustENM to provide implementations for other hybrid methods.
It is tested with a new hybrid method built around adaptive ANM, which works successfully. The following test code produces a transition from 1akeA to 4akeA as confirmed by the plots below and visualisation of the output (not shown):
import matplotlib.pyplot as plt
plt.ion()
from prody import *
r = parsePDB('1akeA')
t = parsePDB('4akeA')
self = AdaptiveHybrid('test')
self.setAtoms(r, t)
self.run(n_gens=20)
plt.figure()
rmsd_from_start = self.getRMSDs()
plt.plot(rmsd_from_start)
plt.savefig('ake_adaptiveHybrid_RMSDs_A')
plt.close()
plt.figure()
rmsd_from_start = self.getRMSDsB()
plt.plot(rmsd_from_start)
plt.savefig('ake_adaptiveHybrid_RMSDs_B')
plt.close()


We can now run ClustENM and then filter to only keep conformers that have the same or better CCC to a target map than a reference structure (this follows MDeNM-EMFit):
import matplotlib.pyplot as plt
plt.ion()
from prody import *
from TEMPy.protein.structure_blurrer import StructureBlurrer
o = parsePDB('1gruA')
c = parsePDB('1gr5A')
proc = 2
if proc == 1:
ens = ClustENM('test')
ens.setAtoms(o)
ens.run(n_gens=3, maxclust=(10, 30, 50))
saveEnsemble(ens, '1gruA_clustenm.ens.npz')
writePDB('1gruA_clustenm.pdb', ens)
else:
ens = loadEnsemble('1gruA_clustenm.ens.npz')
atoms = ens.getAtoms()
amap = alignChains(atoms, o.ca)[0]
ens.setAtoms(amap)
mean_conf = amap.copy()
mean_conf.setCoords(ens.getCoords())
plt.figure()
rmsd_from_start = ens.getRMSDs()
plt.plot(rmsd_from_start)
plt.savefig('1gruA_clustenm_RMSDs')
plt.close()
cmap = alignChains(c, mean_conf)[0]
c_alg, T = superpose(cmap, mean_conf, weights=cmap.getFlags('mapped'))
rmsd_from_cmap = [calcRMSD(coordset, cmap, weights=cmap.getFlags('mapped'))
for coordset in ens.getCoordsets()]
plt.figure()
plt.plot(rmsd_from_cmap)
plt.savefig('1gruA_clustenm_RMSDs_from_1gr5A')
plt.close()
blurrer = StructureBlurrer()
c_Structure = c.toTEMPyStructure()
c_blurred = blurrer.gaussian_blur(c_Structure, 5,
filename=c.getTitle()+'.mrc')
c_blurred.write_to_MRC_file(c_blurred.filename)
defvec_to_cmap = calcDeformVector(mean_conf, cmap,
weights=cmap.getFlags('mapped'))
writeNMD('1gruA_to_1gr5A.defvec.nmd', defvec_to_cmap, mean_conf)
omap = alignChains(o, mean_conf)[0]
o_alg, T = superpose(omap, mean_conf, weights=cmap.getFlags('mapped'))
rmsd_from_omap = [calcRMSD(coordset, omap, weights=omap.getFlags('mapped'))
for coordset in ens.getCoordsets()]
plt.figure()
plt.plot(rmsd_from_omap)
plt.savefig('1gruA_clustenm_RMSDs_from_1gruA')
plt.close()
pca = PCA('1gruA ClustENM')
pca.buildCovariance(ens)
pca.calcModes()
saveModel(pca, '1gruA_ClustENM.pca.npz')
writeNMD('1gruA_ClustENM.pca.nmd', pca, mean_conf)
plt.figure()
showProjection(ens, pca[:2], label='ClustENM')
showProjection(ens[0], pca[:2], c='c', markersize=10, label='1gruA (start)')
showProjection(defvec_to_cmap, pca[:2], c='r', markersize=10, label='1gr5A')
plt.legend()
plt.savefig('1gruA_clustenm_pca_proj2d')
xlim = plt.xlim()
ylim = plt.ylim()
plt.close()
ens.setAtoms(atoms)
ens.filterConformers(ref=o, target=c_blurred)
ens.setAtoms(amap)
plt.figure()
showProjection(ens, pca[:2], label='ClustENM')
showProjection(ens[0], pca[:2], c='c', markersize=10, label='1gruA (start)')
showProjection(defvec_to_cmap, pca[:2], c='r', markersize=10, label='1gr5A')
plt.legend()
plt.xlim(xlim)
plt.ylim(ylim)
plt.savefig('1gruA_clustenm_filtered_pca_proj2d')
plt.close()
ens.setAtoms(atoms)
This can be seen on the PCA projections:


There is now a function calcANMMC to perform a single ANM MC cycle from CoMD:
from prody import *
o = parsePDB('1gruA')
c = parsePDB('1gr5A')
omap = alignChains(o, c.ca)[0]
c_alg, T = superpose(c.ca, omap, weights=omap.getFlags('mapped'))
o_sel = o.select(omap.getSelstr())
cmap = alignChains(c_alg, o_sel)[0]
ens, count1, count2, count3, k, accept_para = calcANMMC(o_sel, cmap)
The outputs are as follows:
@> PDB file is found in working directory (1gru.pdb.gz).
@> 3809 atoms and 1 coordinate set(s) were parsed in 0.18s.
@> Secondary structures were assigned to 372 residues.
@> PDB file is found in working directory (1gr5.pdb.gz).
@> 3762 atoms and 1 coordinate set(s) were parsed in 0.18s.
@> Secondary structures were assigned to 376 residues.
@> Trying to map atoms based on residue numbers and identities:
@> Comparing Chain A from 1gruA (len=525) with Chain A from 1gr5A:
@> Mapped: 517 residues match with 100% sequence identity and 100% overlap.
@> Finding the atommaps based on their coverages...
@> Identified that there exists 1 atommap(s) potentially.
@> Trying to map atoms based on residue numbers and identities:
@> Comparing Chain A from 1gr5A (len=517) with Chain A from 1gruA:
@> Mapped: 516 residues match with 100% sequence identity and 100% overlap.
@> Finding the atommaps based on their coverages...
@> Identified that there exists 1 atommap(s) potentially.
@> Hessian was built in 0.31s.
@> 20 modes were calculated in 0.17s.
rmsd rand ID step accept_para f
0.17 0.44 3 0 1 0.1000 1.0000
0.17 0.12 0 1 0 0.1000 0.0000
0.21 0.58 6 2 1 0.1000 0.0000
0.37 0.41 3 3 1 0.1000 0.0000
0.37 0.68 8 4 0 0.1000 0.0000
0.54 0.45 3 5 1 0.1500 0.0000
0.54 0.32 2 6 0 0.1500 0.0000
0.71 0.46 3 7 1 0.1500 0.0000
0.72 0.70 9 8 1 0.1500 0.0000
0.72 0.28 1 9 0 0.1500 0.0000
0.72 0.32 2 10 0 0.2250 0.0000
0.72 0.70 9 11 0 0.2250 0.0000
0.72 0.31 2 12 0 0.2250 0.0000
0.72 0.55 5 13 0 0.2250 0.0000
0.72 0.89 15 14 1 0.2250 0.0000
0.72 0.79 11 15 1 0.3375 0.0000
0.72 0.10 0 16 0 0.3375 0.0000
0.74 0.79 11 17 1 0.3375 0.0000
0.89 0.01 0 18 1 0.3375 0.0000
0.90 0.88 15 19 1 0.3375 0.0000
0.91 0.56 5 20 1 0.5063 0.0000
0.91 0.23 1 21 0 0.5063 0.0000
0.91 0.54 5 22 0 0.5063 0.0000
0.91 0.48 4 23 0 0.5063 0.0000
0.91 0.25 1 24 0 0.5063 0.0000
1.26 0.12 0 25 1 0.7594 0.0000
1.29 0.34 2 26 1 0.7594 0.0000
1.29 0.14 0 27 0 0.7594 0.0000
1.29 0.58 5 28 0 0.7594 0.0000
1.29 0.31 2 29 0 0.7594 0.0000
1.29 0.04 0 30 0 1.1391 0.0000
1.29 0.95 18 31 0 1.1391 0.0000
1.29 0.44 3 32 0 1.1391 0.0000
1.30 0.62 7 33 1 1.1391 0.0000
1.30 0.96 18 34 0 1.1391 0.0000
1.30 0.89 15 35 0 1.7086 0.0000
1.30 0.67 8 36 1 1.7086 0.0000
1.30 0.31 2 37 0 1.7086 0.0000
1.30 0.65 7 38 0 1.7086 0.0000
1.35 0.29 1 39 1 1.7086 0.0000
1.37 0.66 7 40 1 2.5629 0.0000
1.37 0.77 11 41 0 2.5629 0.0000
1.37 0.96 18 42 0 2.5629 0.0000
1.38 0.68 8 43 1 2.5629 0.0000
1.38 0.81 12 44 0 2.5629 0.0000
1.38 0.23 1 45 0 3.8443 0.0000
1.38 0.16 0 46 0 3.8443 0.0000
1.38 0.66 7 47 0 3.8443 0.0000
1.52 0.22 1 48 1 3.8443 0.0000
1.52 0.03 0 49 0 3.8443 0.0000
1.52 0.51 4 50 0 5.7665 0.0000
1.54 0.68 8 51 1 5.7665 0.0000
1.54 0.68 8 52 0 5.7665 0.0000
1.54 0.70 8 53 0 5.7665 0.0000
1.54 0.85 13 54 0 5.7665 0.0000
1.54 0.87 14 55 0 8.6498 0.0000
1.90 0.01 0 56 1 8.6498 0.0000
2.32 0.13 0 57 1 8.6498 0.0000
It indeed brings the RMSD to the target down as well as bringing the RMSD from the initial structure up:
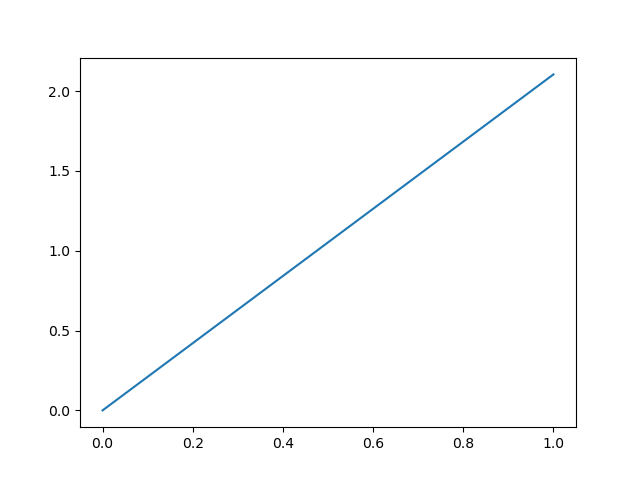
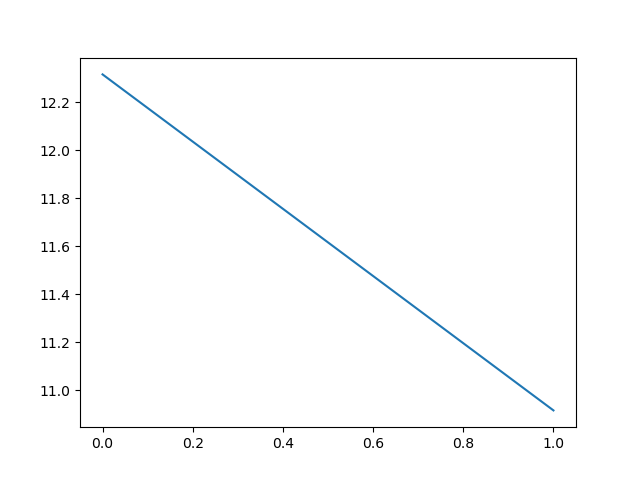
It also works to run it from the command line, like it is used in the original CoMD implementation:
$ python ~/code/ProDy/prody/dynamics/comd.py 1akeA_fixed.pdb 4akeA_fixed.pdb 1ake.pdb.gz 4ake.pdb.gz 1 0.1 1.5
@> 3341 atoms and 1 coordinate set(s) were parsed in 0.06s.
@> 3341 atoms and 1 coordinate set(s) were parsed in 0.07s.
@> Hessian was built in 0.13s.
@> 20 modes were calculated in 0.06s.
rmsd rand ID step accept_para f
0.05 0.77 14 0 1 2.5600 1.0000
0.05 0.58 9 1 0 2.5600 0.0000
0.09 0.17 2 2 1 2.5600 0.0000
0.12 0.28 4 3 1 2.5600 0.0000
0.13 0.65 11 4 1 2.5600 0.0000
0.18 0.33 4 5 1 3.8400 0.0000
0.24 0.31 4 6 1 3.8400 0.0000
0.24 0.30 4 7 0 3.8400 0.0000
0.24 0.88 16 8 0 3.8400 0.0000
0.25 0.26 3 9 1 3.8400 0.0000
0.25 0.10 1 10 0 5.7600 0.0000
0.27 0.05 0 11 1 5.7600 0.0000
0.27 0.76 13 12 1 5.7600 0.0000
0.32 0.01 0 13 1 5.7600 0.0000
0.34 0.76 14 14 1 5.7600 0.0000
0.35 0.65 11 15 1 8.6400 0.0000
0.37 0.26 3 16 1 8.6400 0.0000
0.38 0.49 7 17 1 8.6400 0.0000
0.39 0.80 14 18 1 8.6400 0.0000
0.44 0.28 4 19 1 8.6400 0.0000
0.44 0.10 1 20 0 12.9600 0.0000
0.46 0.18 2 21 1 12.9600 0.0000
0.47 0.45 7 22 1 12.9600 0.0000
0.47 0.20 2 23 0 12.9600 0.0000
0.50 0.26 3 24 1 12.9600 0.0000
0.50 0.41 6 25 1 19.4400 0.0000
0.53 0.22 2 26 1 19.4400 0.0000
0.55 0.42 6 27 1 19.4400 0.0000
0.55 0.72 13 28 0 19.4400 0.0000
0.55 0.08 0 29 0 19.4400 0.0000
0.55 0.90 17 30 0 29.1600 0.0000
0.58 0.17 2 31 1 29.1600 0.0000
0.58 0.36 5 32 0 29.1600 0.0000
0.63 0.22 2 33 1 29.1600 0.0000
0.63 0.19 2 34 0 29.1600 0.0000
0.63 0.02 0 35 0 43.7400 0.0000
0.63 0.13 1 36 0 43.7400 0.0000
0.63 0.99 19 37 0 43.7400 0.0000
0.68 0.16 2 38 1 43.7400 0.0000
0.72 0.33 4 39 1 43.7400 0.0000
0.73 0.66 11 40 1 65.6100 0.0000
0.73 0.14 1 41 0 65.6100 0.0000
0.73 0.66 11 42 0 65.6100 0.0000
0.73 0.86 16 43 0 65.6100 0.0000
0.73 0.38 5 44 0 65.6100 0.0000
0.74 0.78 14 45 1 98.4150 0.0000
0.75 0.45 7 46 1 98.4150 0.0000
0.75 0.22 3 47 0 98.4150 0.0000
0.75 0.83 15 48 0 98.4150 0.0000
0.75 0.30 4 49 0 98.4150 0.0000
0.75 0.85 16 50 0 147.6225 0.0000
0.75 0.85 16 51 1 147.6225 0.0000
0.75 0.50 8 52 0 147.6225 0.0000
0.75 0.72 13 53 0 147.6225 0.0000
0.75 0.27 3 54 0 147.6225 0.0000
0.79 0.03 0 55 1 221.4338 0.0000
0.79 0.82 15 56 0 221.4338 0.0000
0.79 0.97 19 57 1 221.4338 0.0000
0.79 0.73 13 58 0 221.4338 0.0000
0.79 0.13 1 59 1 221.4338 0.0000
0.84 0.21 2 60 1 332.1506 0.0000
0.84 0.78 14 61 0 332.1506 0.0000
0.84 0.96 18 62 0 332.1506 0.0000
0.84 0.29 4 63 0 332.1506 0.0000
0.84 0.61 10 64 0 332.1506 0.0000
0.85 0.86 16 65 1 498.2259 0.0000
0.85 0.95 18 66 0 498.2259 0.0000
0.85 0.08 1 67 0 498.2259 0.0000
0.85 0.72 13 68 0 498.2259 0.0000
0.85 0.04 0 69 0 498.2259 0.0000
0.85 0.36 5 70 0 747.3389 0.0000
0.85 0.56 9 71 1 747.3389 0.0000
0.85 0.25 3 72 0 747.3389 0.0000
0.85 0.60 10 73 1 747.3389 0.0000
0.85 0.91 17 74 1 747.3389 0.0000
0.85 0.91 17 75 0 1121.0084 0.0000
0.88 0.33 4 76 1 1121.0084 0.0000
0.88 0.49 8 77 0 1121.0084 0.0000
0.88 0.62 10 78 0 1121.0084 0.0000
0.88 0.75 13 79 0 1121.0084 0.0000
0.89 0.92 17 80 1 1681.5125 0.0000
0.89 0.81 15 81 0 1681.5125 0.0000
0.89 0.85 16 82 0 1681.5125 0.0000
0.90 0.45 7 83 1 1681.5125 0.0000
0.90 0.85 16 84 0 1681.5125 0.0000
0.90 0.20 2 85 0 2522.2688 0.0000
0.90 0.39 5 86 0 2522.2688 0.0000
0.90 0.33 4 87 0 2522.2688 0.0000
0.91 0.42 6 88 1 2522.2688 0.0000
0.93 0.13 1 89 1 2522.2688 0.0000
0.93 0.84 15 90 1 3783.4032 0.0000
0.93 0.72 13 91 0 3783.4032 0.0000
0.93 0.95 18 92 0 3783.4032 0.0000
0.93 0.98 19 93 0 3783.4032 0.0000
0.93 0.49 7 94 0 3783.4032 0.0000
0.93 0.67 11 95 0 5675.1048 0.0000
0.93 0.83 15 96 0 5675.1048 0.0000
0.97 0.07 0 97 1 5675.1048 0.0000
0.97 0.16 2 98 0 5675.1048 0.0000
0.97 0.85 16 99 0 5675.1048 0.0000
0.99 0.27 3 100 1 8512.6572 0.0000
0.99 0.01 0 101 0 8512.6572 0.0000
1.03 0.03 0 102 1 8512.6572 0.0000
1.03 0.75 13 103 0 8512.6572 0.0000
1.03 0.04 0 104 0 8512.6572 0.0000
1.03 0.37 5 105 1 12768.9858 0.0000
1.03 0.73 13 106 0 12768.9858 0.0000
1.04 0.86 16 107 1 12768.9858 0.0000
1.04 0.65 11 108 0 12768.9858 0.0000
1.05 0.42 6 109 1 12768.9858 0.0000
1.06 0.52 8 110 1 19153.4788 0.0000
1.06 0.67 11 111 0 19153.4788 0.0000
1.11 0.07 0 112 1 19153.4788 0.0000
1.11 0.80 15 113 0 19153.4788 0.0000
1.11 0.34 5 114 1 19153.4788 0.0000
1.11 0.02 0 115 0 28730.2181 0.0000
1.11 0.32 4 116 0 28730.2181 0.0000
1.11 0.42 6 117 0 28730.2181 0.0000
1.12 0.84 16 118 1 28730.2181 0.0000
1.15 0.29 4 119 1 28730.2181 0.0000
1.15 0.57 9 120 1 43095.3272 0.0000
1.15 0.44 7 121 0 43095.3272 0.0000
1.15 0.70 12 122 1 43095.3272 0.0000
1.19 0.18 2 123 1 43095.3272 0.0000
1.21 0.12 1 124 1 43095.3272 0.0000
1.22 0.61 10 125 1 64642.9908 0.0000
1.22 0.68 12 126 0 64642.9908 0.0000
1.22 0.91 17 127 0 64642.9908 0.0000
1.22 0.79 14 128 0 64642.9908 0.0000
1.22 0.09 1 129 0 64642.9908 0.0000
1.22 0.64 11 130 0 96964.4862 0.0000
1.22 0.62 10 131 0 96964.4862 0.0000
1.22 0.76 14 132 0 96964.4862 0.0000
1.22 0.63 11 133 0 96964.4862 0.0000
1.22 0.02 0 134 0 96964.4862 0.0000
1.22 0.68 12 135 0 145446.7294 0.0000
1.23 0.46 7 136 1 145446.7294 0.0000
1.23 0.69 12 137 0 145446.7294 0.0000
1.23 0.94 18 138 0 145446.7294 0.0000
1.25 0.10 1 139 1 145446.7294 0.0000
1.25 0.57 9 140 0 218170.0941 0.0000
1.27 0.26 3 141 1 218170.0941 0.0000
1.27 0.89 17 142 0 218170.0941 0.0000
1.27 0.01 0 143 0 218170.0941 0.0000
1.32 0.22 2 144 1 218170.0941 0.0000
1.32 0.70 12 145 1 327255.1411 0.0000
1.32 0.98 19 146 0 327255.1411 0.0000
1.32 0.90 17 147 1 327255.1411 0.0000
1.32 0.36 5 148 0 327255.1411 0.0000
1.32 0.88 16 149 0 327255.1411 0.0000
1.32 0.53 8 150 0 490882.7116 0.0000
1.33 0.83 15 151 1 490882.7116 0.0000
1.33 0.87 16 152 0 490882.7116 0.0000
1.36 0.11 1 153 1 490882.7116 0.0000
1.39 0.12 1 154 1 490882.7116 0.0000
1.39 0.49 8 155 1 736324.0675 0.0000
1.39 0.03 0 156 0 736324.0675 0.0000
1.40 0.70 12 157 1 736324.0675 0.0000
1.40 0.90 17 158 1 736324.0675 0.0000
1.41 0.59 10 159 1 736324.0675 0.0000
1.42 0.77 14 160 1 1104486.1012 0.0000
1.42 0.24 3 161 0 1104486.1012 0.0000
1.42 0.82 15 162 0 1104486.1012 0.0000
1.46 0.13 1 163 1 1104486.1012 0.0000
1.48 0.22 3 164 1 1104486.1012 0.0000
1.48 0.57 9 165 0 1656729.1518 0.0000
1.49 0.35 5 166 1 1656729.1518 0.0000
1.49 0.78 14 167 0 1656729.1518 0.0000
1.49 0.98 19 168 1 1656729.1518 0.0000
1.49 0.05 0 169 0 1656729.1518 0.0000
1.53 0.19 2 170 1 2485093.7277 0.0000
@> DCD file was written in 0.00 seconds.
@> 0.00 MB written at input rate 1.26 MB/s.
@> 1 coordinate sets written at output rate 500 frame/s.
This was the original error when providing fewer than 3 input arguments:
$ python ~/code/ProDy/prody/dynamics/comd.py 1gru_A.pdb 1gr5_A.pdb
Traceback (most recent call last):
File "C:/Users/james/code/ProDy/prody/dynamics/comd.py", line 197, in <module>
original_initial_pdb = ar[3]
IndexError: list index out of range
Now, it allows the user to provide just 1 input argument (a PDB filename) and raises a more sensible error if it doesn't get one:
$ python ~/code/ProDy/prody/dynamics/comd.py
Traceback (most recent call last):
File "C:/Users/james/code/ProDy/prody/dynamics/comd.py", line 195, in <module>
raise ValueError('Please provide at least 1 argument (a PDB filename)')
ValueError: Please provide at least 1 argument (a PDB filename)
It also autofills the remaining arguments based on the ones it gets. If it gets just 1 filename, then it fills in the initial and final filenames and the original ones with those names and knows that it should work in exploration mode. If it gets 2 then it fills the initial and final filenames with those and uses the same ones for the original initial and final filenames so it can check if it should be in exploration or transition mode. It can also do this for just the second one of those. Finally, it also autofills the cycle number as 1 if not given one.
We now have a working version of CoMD.
Running code:
from prody import *
import matplotlib.pyplot as plt
plt.ion()
o = parsePDB('4akeA')
c = parsePDB('1akeA')
self = CoMD('ake')
self.setAtoms(o, c)
self.run(n_gens=20)
plt.figure()
rmsd_from_start = self.getRMSDs()
plt.plot(rmsd_from_start, 'o-')
plt.savefig('ake_comd_RMSDs_A')
plt.close()
plt.figure()
rmsd_from_target = self.getRMSDsB()
plt.plot(rmsd_from_target, 'o-')
plt.savefig('ake_comd_RMSDs_B')
plt.close()
plt.figure()
rmsd_mat = self.getRMSDs(pairwise=True)
showMatrix(rmsd_mat)
plt.savefig('ake_comd_RMSD_matrix')
plt.close()
Output start:
@> PDB file is found in working directory (4ake.pdb.gz).
@> 1728 atoms and 1 coordinate set(s) were parsed in 0.02s.
@> Secondary structures were assigned to 139 residues.
@> PDB file is found in working directory (1ake.pdb.gz).
@> 1954 atoms and 1 coordinate set(s) were parsed in 0.03s.
@> Secondary structures were assigned to 153 residues.
@> Fixing structure A...
@> 3341 atoms and 1 coordinate set(s) were parsed in 0.04s.
@> The structure was fixed in 2.93s.
@> Fixing structure B...
@> 3341 atoms and 1 coordinate set(s) were parsed in 0.04s.
@> The structure was fixed in 1.54s.
@> Kirchhoff was built in 0.02s.
@> Generation 0 for structure A ...
@> Minimization in generation 0 for structure A ...
@> Completed in 0.82s.
@> #-------------------/*\-------------------#
@> Kirchhoff was built in 0.02s.
@> Generation 0 for structure B ...
@> Minimization in generation 0 for structure B ...
@> Completed in 0.78s.
@> #-------------------/*\-------------------#
@> Generation 1 ...
@> Sampling conformers in generation 1 ...
@> Hessian was built in 0.07s.
@> Hessian was built in 0.10s.
@>
Starting cycle with structure A
@> Hessian was built in 0.11s.
@> 20 modes were calculated in 0.02s.
rmsd rand ID step accept_para f
0.22 0.43 2 0 1 0.1000 1.0000
0.23 0.97 18 1 1 0.1000 1.0000
0.23 0.14 0 2 0 0.1000 0.0000
0.23 0.52 4 3 0 0.1000 0.0000
0.23 0.93 16 4 0 0.1000 0.0000
0.23 0.41 2 5 0 0.1500 0.0000
0.23 0.65 7 6 0 0.1500 0.0000
0.55 0.12 0 7 1 0.1500 0.0000
0.55 0.43 2 8 0 0.1500 0.0000
0.55 0.32 1 9 0 0.1500 0.0000
0.55 0.82 12 10 1 0.2250 0.0000
0.55 0.36 2 11 0 0.2250 0.0000
1.03 0.09 0 12 1 0.2250 0.0000
@> Parameter: rmsd = 1.03 A
@> Parameter: n_steps = 1
@> Step size is 1.03 A RMSD
@> Mode is scaled by 59.370579906575706.
@> RMSD: 1.03 -> 0.37
@> 1/1 sets of coordinates were moved to the target
@> Structures were sampled in 0.68s.
@> #-------------------/*\-------------------#
C:\Users\james\code\ProDy\prody\dynamics\hybrid.py:554: RuntimeWarning: invalid value encountered in true_divide
tmp = 0.6745 * (arg - np.median(arg)) / mad(arg)
@> Generation 2 ...
@> Sampling conformers in generation 2 ...
@> Hessian was built in 0.07s.
@> Hessian was built in 0.09s.
@>
Starting cycle with structure B
@> Hessian was built in 0.09s.
@> 20 modes were calculated in 0.02s.
rmsd rand ID step accept_para f
0.00 0.37 5 0 0 0.1000 0.0000
0.30 0.50 8 1 1 0.1000 0.0000
0.44 0.39 6 2 1 0.1000 0.0000
0.44 0.72 13 3 0 0.1000 0.0000
0.44 0.27 3 4 0 0.1000 0.0000
0.44 0.60 10 5 0 0.1500 0.0000
0.44 0.82 15 6 0 0.1500 0.0000
0.58 0.28 3 7 1 0.1500 0.0000
0.68 0.33 4 8 1 0.1500 0.0000
0.82 0.15 1 9 1 0.1500 0.0000
0.86 0.64 11 10 1 0.2250 0.0000
0.86 0.78 14 11 0 0.2250 0.0000
0.86 0.31 4 12 0 0.2250 0.0000
0.86 0.46 7 13 0 0.2250 0.0000
0.86 0.00 0 14 0 0.2250 0.0000
1.05 0.32 4 15 1 0.3375 0.0000
@> Parameter: rmsd = 1.05 A
@> Parameter: n_steps = 1
@> Step size is 1.05 A RMSD
@> Mode is scaled by 60.94961805844272.
@> RMSD: 1.05 -> 0.53
@> 1/1 sets of coordinates were moved to the target
@> Structures were sampled in 0.74s.
@> #-------------------/*\-------------------#
Output final:
Starting cycle with structure A
@> Hessian was built in 0.08s.
@> 20 modes were calculated in 0.02s.
rmsd rand ID step accept_para f
0.12 0.80 13 0 1 0.1000 1.0000
0.33 0.25 2 1 1 0.1000 1.0000
0.35 0.71 10 2 1 0.1000 1.0000
0.35 0.56 7 3 0 0.1000 0.0000
0.35 0.53 6 4 0 0.1000 0.0000
0.35 0.61 8 5 0 0.1500 0.0000
0.35 0.76 12 6 0 0.1500 0.0000
0.35 0.36 3 7 0 0.1500 0.0000
0.45 0.39 3 8 1 0.1500 0.0000
0.48 0.55 6 9 1 0.1500 0.0000
0.48 0.81 13 10 0 0.2250 0.0000
0.48 0.02 0 11 0 0.2250 0.0000
0.48 0.09 0 12 0 0.2250 0.0000
0.48 0.48 5 13 0 0.2250 0.0000
0.48 0.60 7 14 0 0.2250 0.0000
0.48 0.28 2 15 0 0.3375 0.0000
0.48 0.70 10 16 0 0.3375 0.0000
0.57 0.53 6 17 1 0.3375 0.0000
0.58 0.87 15 18 1 0.3375 0.0000
0.58 0.48 5 19 0 0.3375 0.0000
0.58 0.43 4 20 0 0.5063 0.0000
0.78 0.27 2 21 1 0.5063 0.0000
0.79 0.99 19 22 1 0.5063 0.0000
0.79 0.50 5 23 0 0.5063 0.0000
0.89 0.16 1 24 1 0.5063 0.0000
0.91 0.98 19 25 1 0.7594 0.0000
1.03 0.35 3 26 1 0.7594 0.0000
@> Parameter: rmsd = 1.03 A
@> Parameter: n_steps = 1
@> Step size is 1.03 A RMSD
@> Mode is scaled by 59.318064026395646.
@> RMSD: 1.03 -> 0.38
@> 1/1 sets of coordinates were moved to the target
@> Structures were sampled in 0.65s.
@> #-------------------/*\-------------------#
@> Generation 10 ...
@> Sampling conformers in generation 10 ...
@> Hessian was built in 0.07s.
@> Hessian was built in 0.09s.
@>
Starting cycle with structure B
@> Hessian was built in 0.10s.
@> 20 modes were calculated in 0.02s.
rmsd rand ID step accept_para f
0.00 0.40 5 0 0 0.1000 0.0000
0.00 0.10 1 1 0 0.1000 0.0000
0.00 0.65 11 2 0 0.1000 0.0000
0.30 0.25 3 3 1 0.1000 0.0000
0.30 0.95 18 4 0 0.1000 0.0000
0.36 0.96 18 5 1 0.1500 0.0000
0.63 0.25 3 6 1 0.1500 0.0000
0.63 0.17 1 7 0 0.1500 0.0000
0.63 0.79 14 8 0 0.1500 0.0000
0.63 0.47 7 9 0 0.1500 0.0000
0.63 0.56 9 10 0 0.2250 0.0000
0.63 0.51 8 11 0 0.2250 0.0000
0.63 0.15 1 12 0 0.2250 0.0000
0.63 0.76 13 13 0 0.2250 0.0000
0.63 0.53 8 14 0 0.2250 0.0000
0.67 0.74 13 15 1 0.3375 0.0000
0.75 0.21 2 16 1 0.3375 0.0000
0.75 0.00 0 17 0 0.3375 0.0000
0.75 0.80 14 18 0 0.3375 0.0000
0.90 0.01 0 19 1 0.3375 0.0000
0.90 0.66 11 20 0 0.5063 0.0000
0.93 0.85 16 21 1 0.5063 0.0000
0.93 0.60 10 22 0 0.5063 0.0000
0.93 0.62 10 23 0 0.5063 0.0000
0.96 0.49 7 24 1 0.5063 0.0000
0.96 0.48 7 25 0 0.7594 0.0000
0.96 0.88 16 26 0 0.7594 0.0000
0.94 0.73 13 27 1 0.7594 0.0000
0.94 0.82 15 28 0 0.7594 0.0000
0.97 0.43 6 29 1 0.7594 0.0000
0.97 0.83 15 30 0 1.1391 0.0000
1.00 0.70 12 31 1 1.1391 0.0000
1.00 0.47 7 32 0 1.1391 0.0000
1.10 0.41 6 33 1 1.1391 0.0000
@> Parameter: rmsd = 1.10 A
@> Parameter: n_steps = 1
@> Step size is 1.10 A RMSD
@> Mode is scaled by 63.40325257043039.
@> RMSD: 1.10 -> 0.48
@> 1/1 sets of coordinates were moved to the target
@> Structures were sampled in 0.65s.
@> #-------------------/*\-------------------#
@> Transition converged in 55.35s.
@>
Ran 10 generations, RMSD 2.8898703631562603, change in RMSD 0.11154516306383844
@> Creating an ensemble of conformers ...
@> Ensemble was created in 0.00s.
@> All completed in 55.35s.
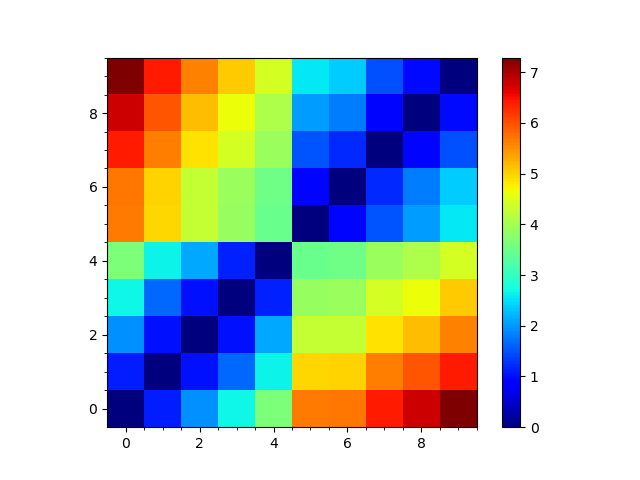
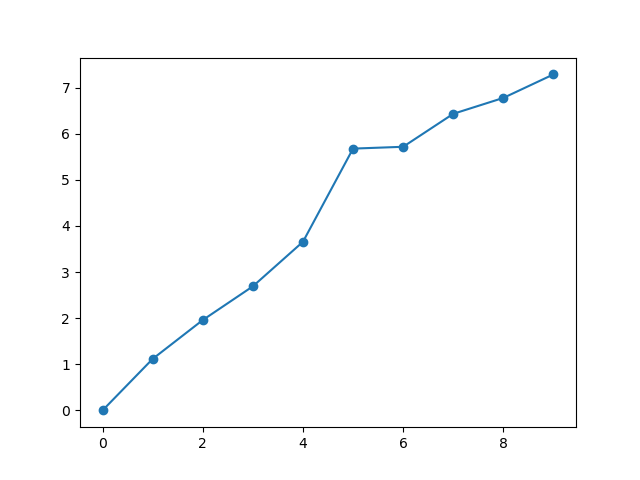
It was previously not adding the conformers to build the ensemble correctly because negative indexing needs to be different depending on whether there are an even or odd number of them.
It could be a good idea to sub/super class AdaptiveHybrid and CoMD so we don't need to keep changing both of them.