Elements and Functional Groups Currently not covered
This a list of elements or functional groups that we currently don't cover with smirnoff99Frosst. I'm not necessarily suggesting we "should" or need to include anything listed here. This is a list of anything that would be assigned generic parameters given the current smirnoff99Frosst (as of 5/2017).
For the Bonds, Angles, and Torsions, I've shown images of the molecules and included parm@frosst atomtypes when available. There is a list of the molecule database we have looked at below.
Elements
These are elements that we do not include and are therefore missing all associated parameters.
- Boron (Discussion in smarty issue)
- Silicon
- Tellurium
- Selenium
- Arsenic
Bonds
From DrugBank Database
5 molecules with the generic bond assigned





- Cl-O
- Cl=O
- O=O
- C#O
- Halogen-Halogen
From Zinc Molecule Set
One molecule with the generic bond assigned

- Cl-O
FreeSolv
No molecules with the generic bond assigned
eMolecules
A note this is not every missing molecules (that's greater than 1,000 molecules, but an idea of the functional groups that were not in the other sets
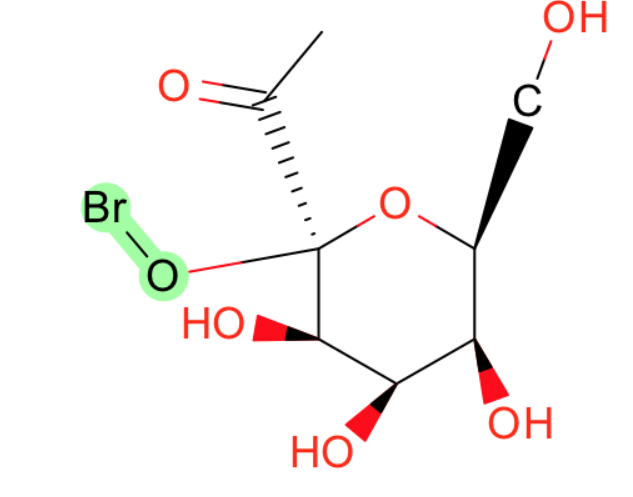
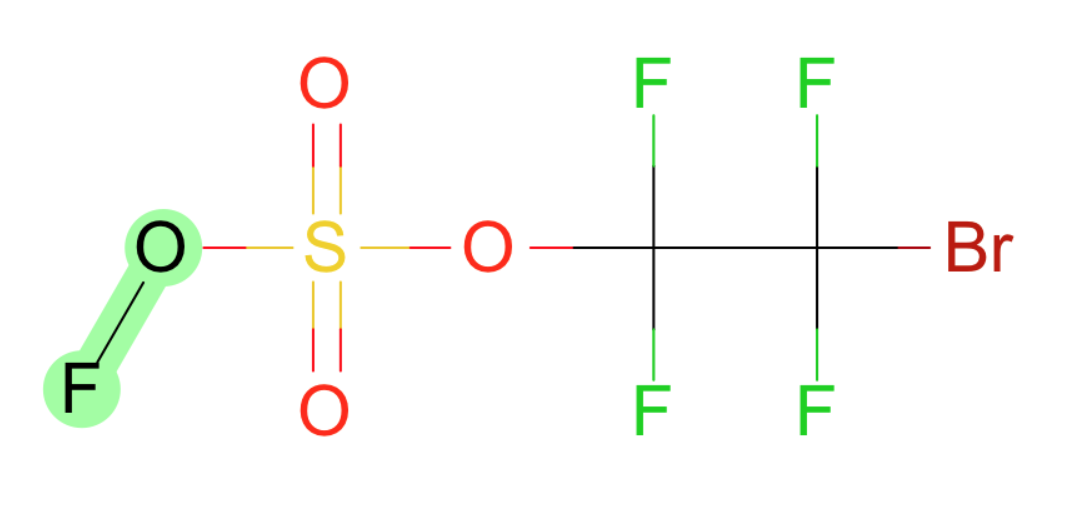
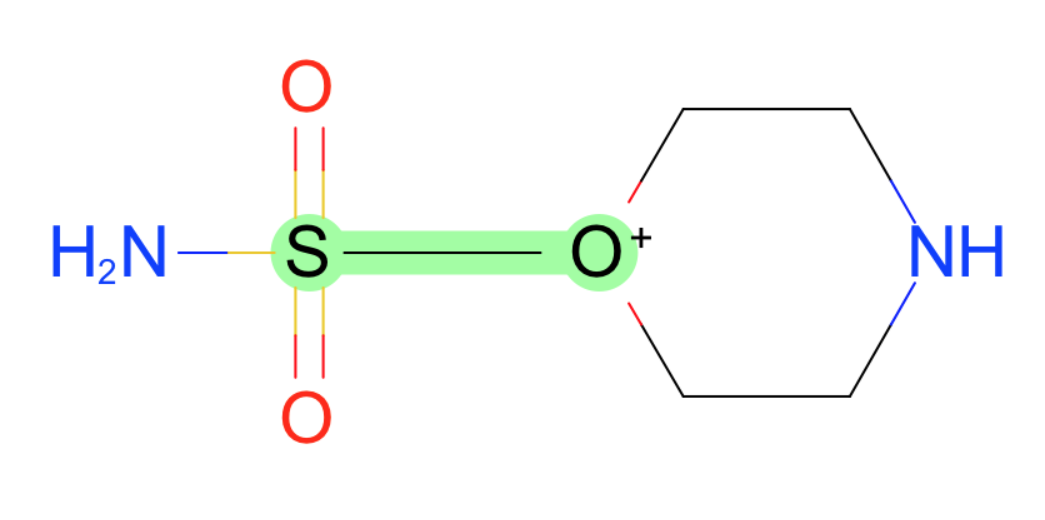
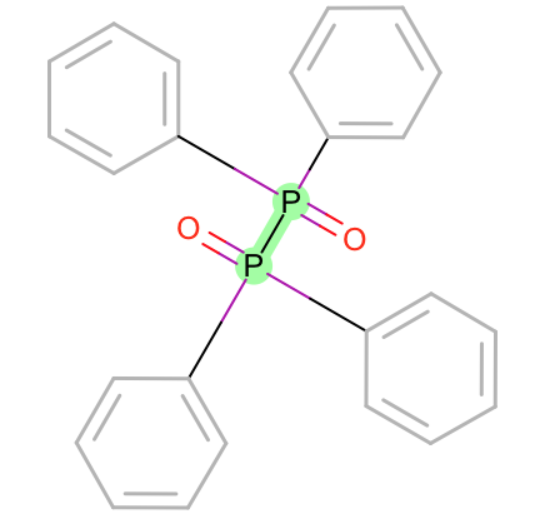
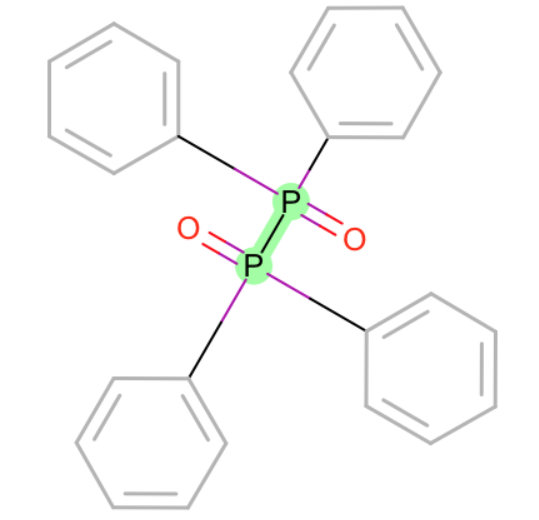
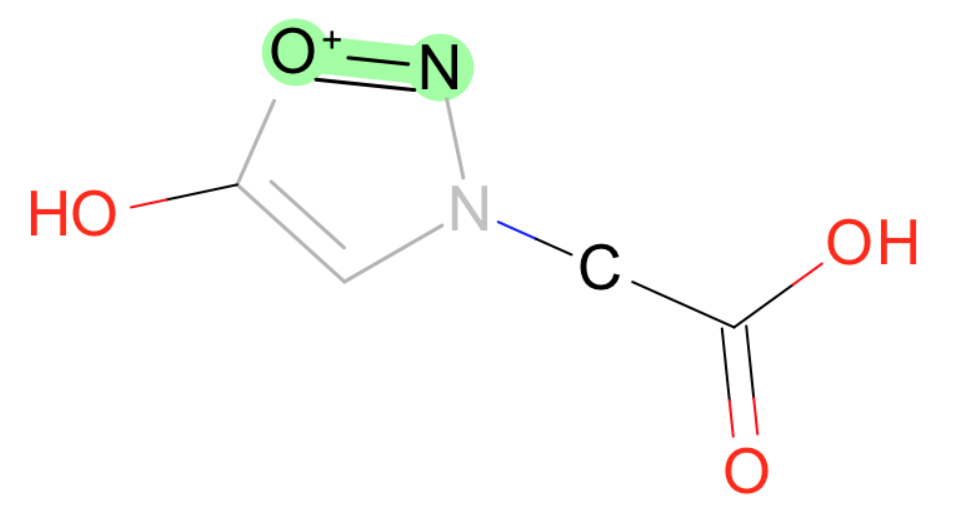
- Br-O
- I-O
- S-[O+1]
- F-O
- P-P
surprising eMolecules
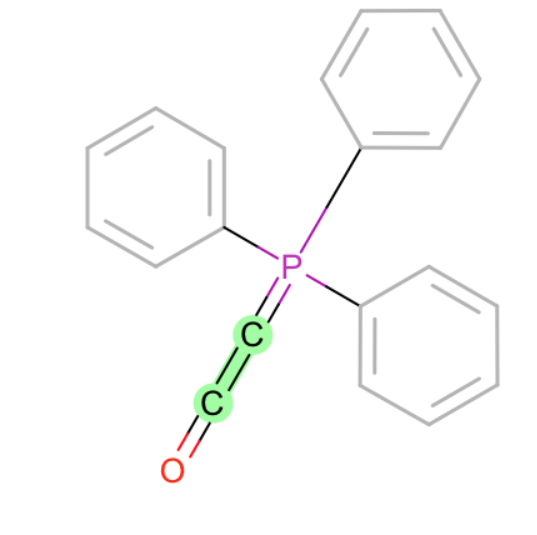
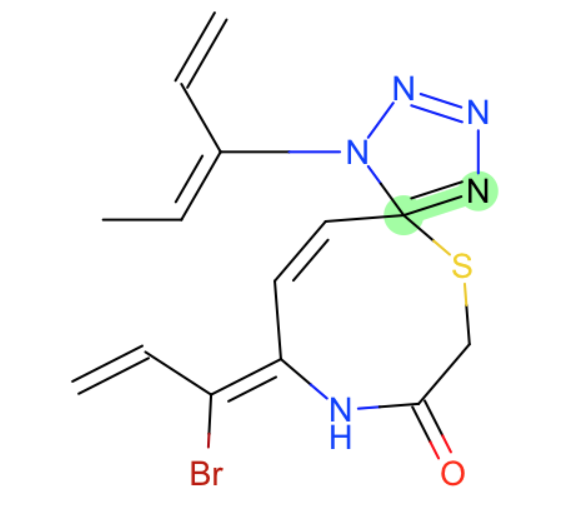
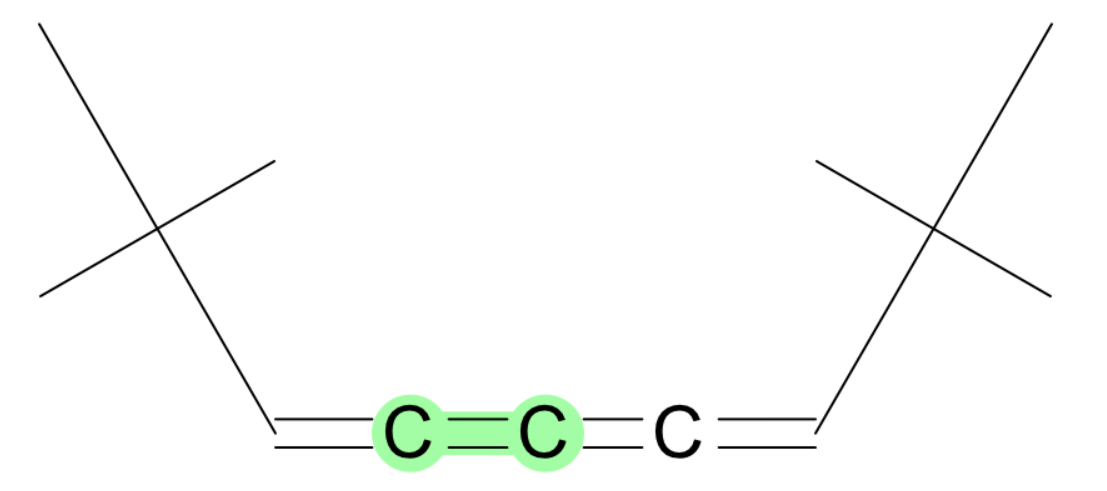
- specific C=N bond
- C=C in chain of double bonds? this seems a bit odd that we miss it, but I imagine these groups may be relatively reactive and therefore not something you typically want to model in drug like molecules.
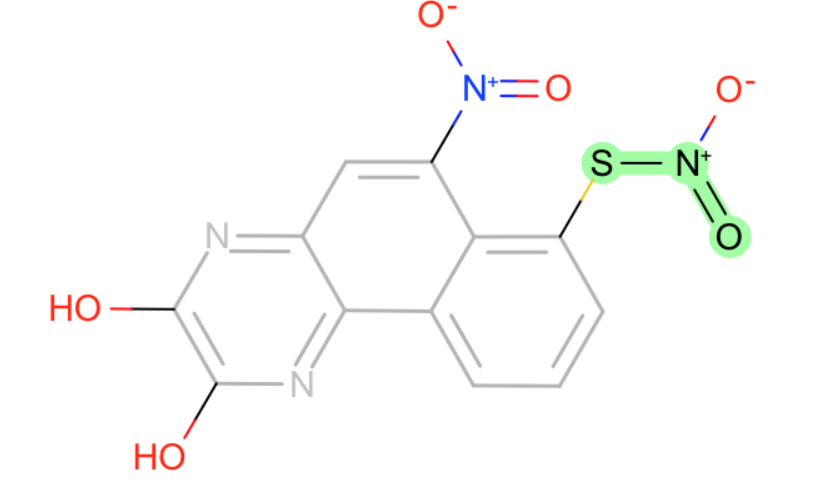
Weird chemistry in eMolecules
- There was more than one molecule with this weird aromatic ring with an extra double bond:
Angles
From DrugBank Database
4 molecules with the generic angle assigned




- C-Cl=O (
CA~CL~Ou) - O=Cl=O (
Ou~CL~Ou) - O-Cl=O (
O2~CL~Ou) - F-S-F (
F~Su~F) - C-N=O (
CT~N3~Ou) - O-N=O (
OH~N3~Ou)- I think this functional group should be a nitrate, I don't trust the nitrogen with four bonds and a radical?
From Zinc Molecule Set
No molecules with the generic angle assigned
FreeSolv
No molecules with the generic angle assigned
eMolecules
These all have weird charged nitrogens.
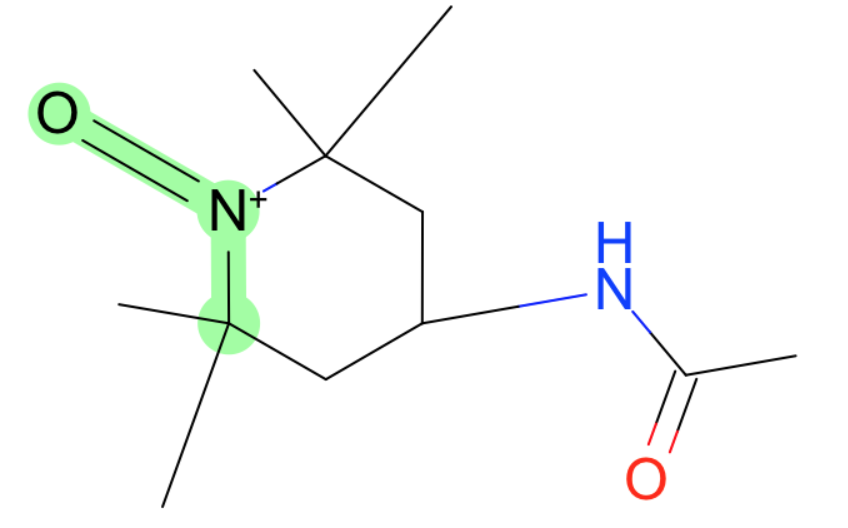
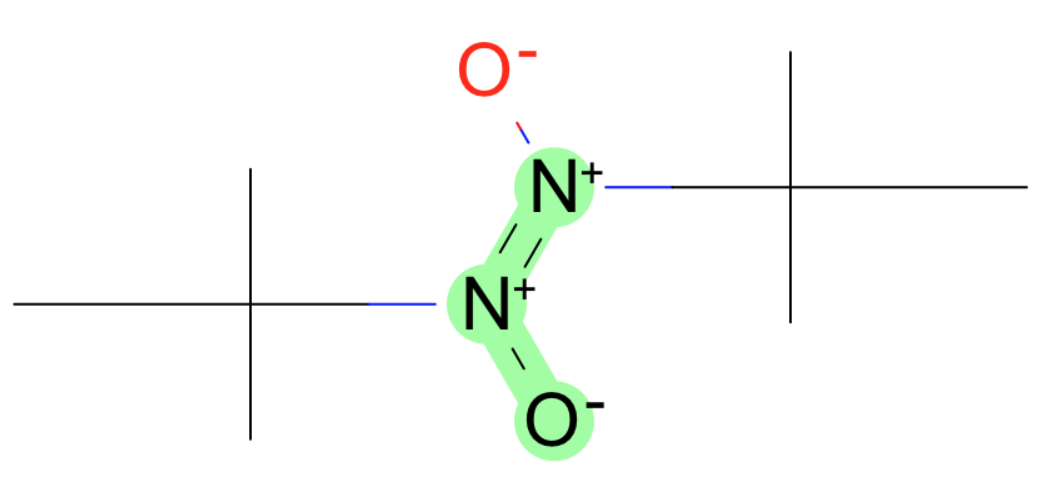
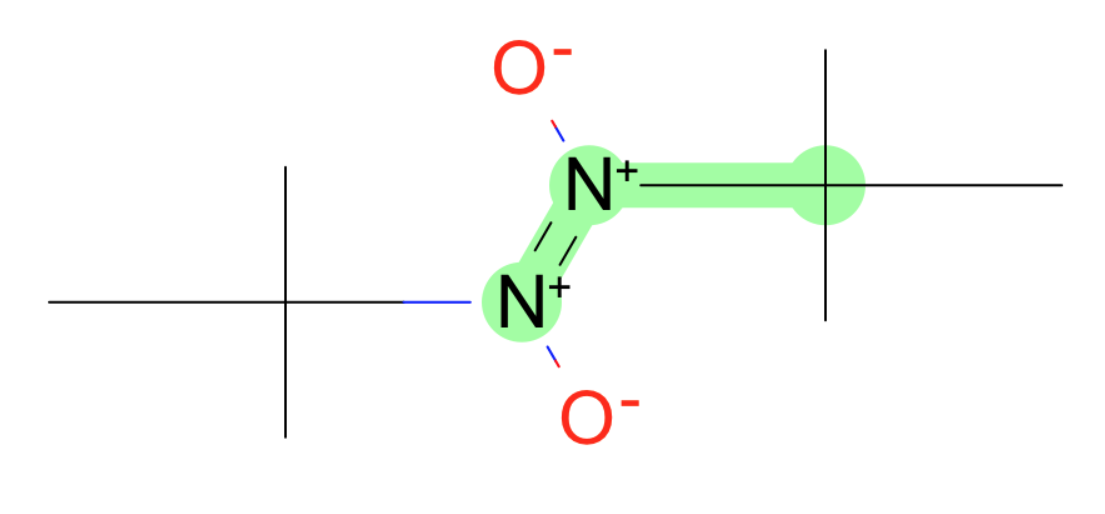
- S-N-O in nitro group
- O=[N+1]-C
- [O-]-[N+]=[N+]
- C-[N+]=[N+] This last one seems like it might be a representation issue, you could also right this functional group as O=N-N-[O-] with less formal charges, but I guess our SMIRKS patterns need to recognize it as the same molecule. Would be worth looking to see if GAFF can type it.
Torsions
From DrugBank Molecule Set
9 molecules with the generic torsion assigned









- [C,H]-N-[nitrate N]~[nitrate O] (
[H,C]~N~N2~O2) - c:c-Cl=O (
[CA,CB]~CA~CL~Ou) - [H,C]-N-S=C (
CT~N3~S~CM) - [C,H]-C-S=O (
[CT,H1]~CT~S~O)- more broadly torsions around X~[C,N]~S=X bonds
- O=C-[O+1]-H (
O~C~Ou~HO) *c:c:,-[SA,Sa;+1]:,-c ([CA,CB]~CB~S~CB)
From Zinc Molecule Set
7 molecules with the generic torsion assigned







- C-N=P-N (
CB~NB~P~NA) - C-P=C-[C,N] (
CC~P~CR~[CT,NA]) - c:c=[O+1]-C (
CA~C~O~CT) - c:c:[O+1]:c (
[CB,CA]~CB~O~CA) - N=[N+1]-C-[C,H] (
NB~NB~CT~[CM,HP,CA,C])
FreeSolv
No molecules with the generic torsion assigned
eMolecules
There were only 948 molecules with "t1" in the old format, that is remarkably small for the 5million+ molecules tested. Most have the P=C or P=N bonds similar to drug bank. Below are some potentially notable additions.
-
*~N-P~ON next to phosphate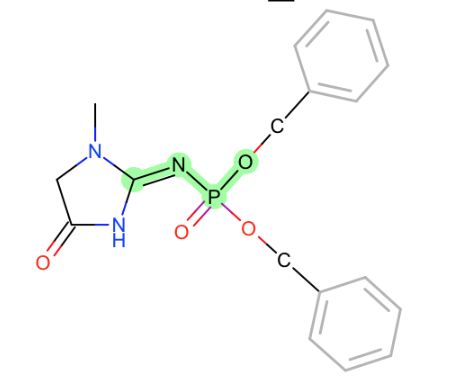
-
this is similar to drugbank in principle, but the neighboring phosphate like groups caught my eye
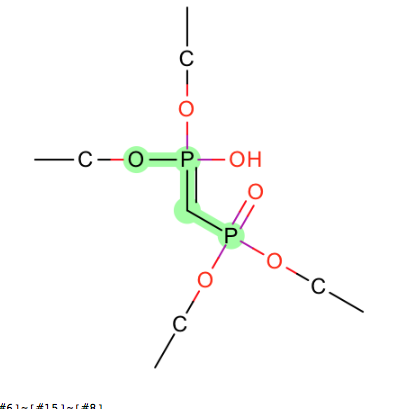
-
C-N-[N+1]=Nsimilar to the missing molecules in zinc
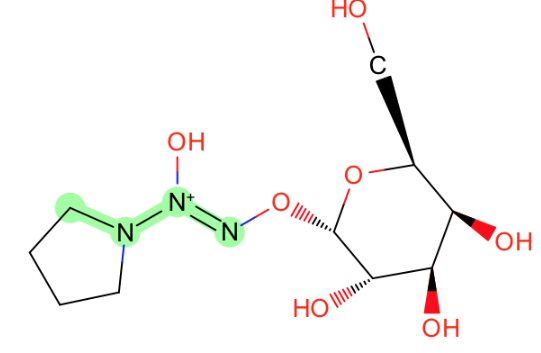
-
sulfur next to phosphorous in those
N-Pbonds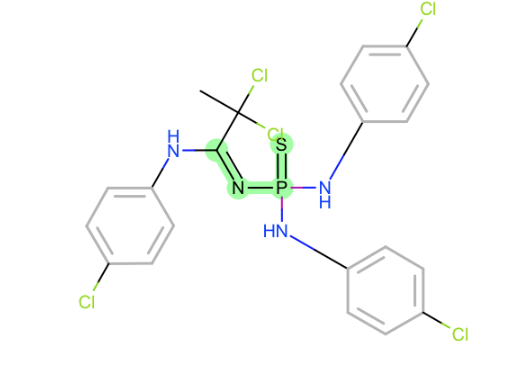
-
C-[N+]=C-Ssimilar to above, but with sulfur, a reminder to keep new parameters general when possible
-
C~P=C-[halogen]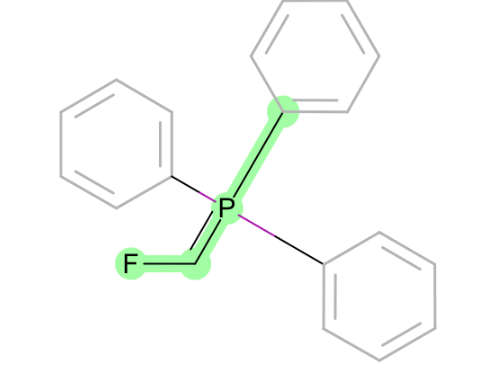
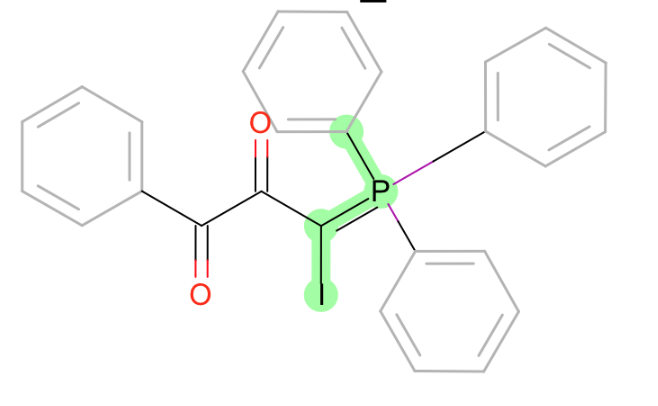
-
S-P weirdness:
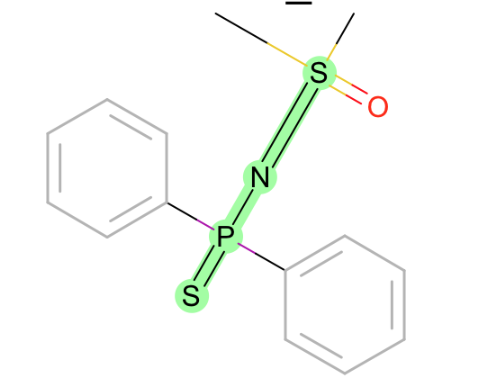
-
all torsions around S-S:
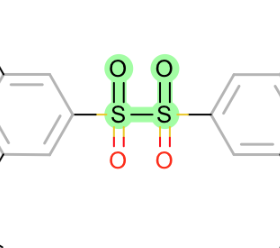
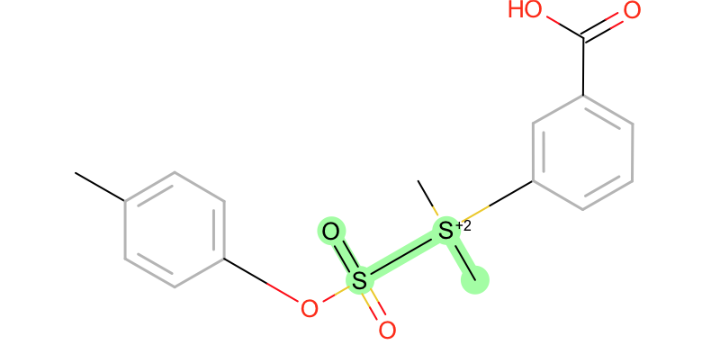
-
extreme version of the
N~Pproblem
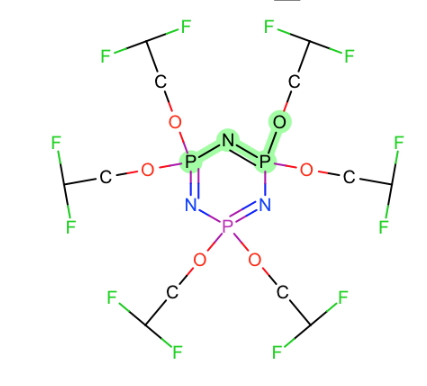
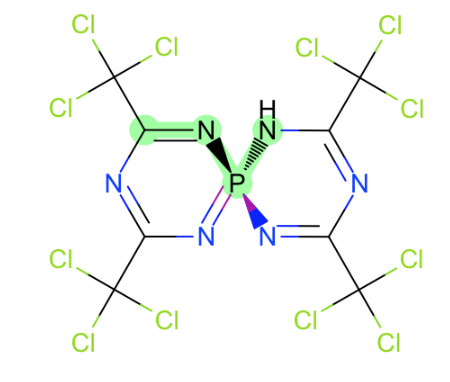
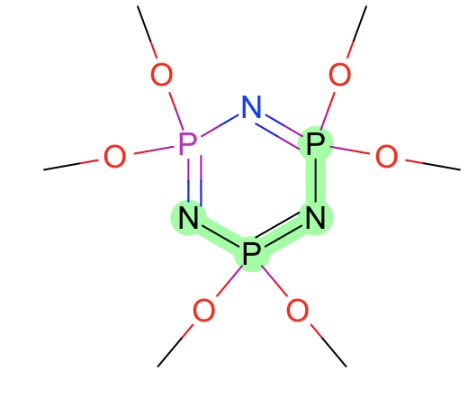
Generally speaking torsions around these bonds:
- Around Nsp2-P and N=P
- Nsp3 was covered with issue #32
- C=P
- C-P
- C=[O+]
- C-[O+]
- S-S
- P-P
Molecule set:
-
openforcefield/data/molecules/DrugBank_atyped.oeb 6647 molecules
- 15/6647 molecules get generic parameters
- Removed 192 molecules with metals
- Removed molecules with boron (56), silicon (4), arsenic (12), selenium(14), tellurium (1), helium(1), and xenon(1).
- Removed 7 molecules with more than 200 heavy atoms
- Removed 11 molecules with smaller atoms (atomic number < 10) with bond order greater than 4
- Removed 66 entries with more than one molecule, that is had a period ('.') in the SMILES string
-
openforcefield/data/molecules/zinc-subset-tripos.mol2.gz 7500 molecules
- 8/7500 molecules assigned a generic parameter
- Removed 5 molecules with smaller atoms (atomic number < 10) with bond order greater than 4
- This database had no metals, non-metals, or entries with multiple molecules
-
MobleyLab/FreeSolv 643
- No filtering
- 0/643 molecules assigned a generic parameter
- eMolecules - I'm currently working on this set, it is MUCH larger (initially 8 million molecules) so there are some logistical barriers to contend with.
- 5,689,262 after filtering for metals, metaloids, and inappropriate valency
- GAFF cannot parameterize 357,589
- 1,036 molecules get generic parameters
I had a reminder in Slack about this article that models a ligand with Boron, potentially a source of data or temporary parameters. Saving here is a better place, it is in the Mobley Group Zotero as well https://pubs.acs.org/doi/10.1021/jacs.6b06566