openff-toolkit
 openff-toolkit copied to clipboard
openff-toolkit copied to clipboard
DrugBank_6182, RDKit, and OpenEye atom stereochemistry assumptions
Describe the bug
DrugBank_6182, when read by RDKitToolkitWrapper, creates a Molecule that when passed to OpenEyeToolkitWrapper's to_openeye() raises the exception "Programming error: OpenEye atom stereochemistry assumptions failed".
This may seem like a "then don't do that" error. However, it also means that repr(rd_mol) fails for that case, even though there is no apparent use of OEChem in the call. The reason is that the Molecule's __repr__ uses the first (default) toolkit to generate the SMILES string for the output, which defaults to OpenEyeToolkitWrapper.
Another place this causes a problem is a check like rd_mol.is_isomorphic_with(rd_mol), because PR #646 added the strip_pyrimidal_n_atom_stereo = True kwarg to is_isomorphic_with(). This calls strip_atom_stereochemistry() which in turn calls chemical_environment_matches on a SMARTS pattern, which (eventually) calls to_openeye() for the default SMARTS matcher.
To Reproduce
import io
import pathlib
import openff.toolkit
from openff.toolkit import utils
# Extract DrugBank_6182 from MiniDrugBank.sdf
mini_drug_bank = pathlib.Path(openff.toolkit.__file__).parent.joinpath(
"data", "molecules", "MiniDrugBank.sdf").read_bytes()
start = mini_drug_bank.find(b"DrugBank_6182")
end = mini_drug_bank.find(b"$$$$\n", start) + 5
DrugBank_6182 = mini_drug_bank[start:end]
# Use RDKitToolkitWrapper to read the record
rd_wrapper = utils.RDKitToolkitWrapper()
f = io.BytesIO(DrugBank_6182)
rd_mol = rd_wrapper.from_file_obj(f, "SDF")[0]
# Attempt to print its repr.
print(repr(rd_mol))
# The previous line raised an exception. So would this line.
rd_mol.is_isomorphic_with(rd_mol))
Output
I've tweaked the exception message to include the failing test:
Traceback (most recent call last):
File "oe_assumptions.py", line 21, in <module>
print(repr(rd_mol))
File "openff/toolkit/topology/molecule.py", line 2611, in __repr__
self.name, self.to_smiles()
File "openff/toolkit/topology/molecule.py", line 2738, in to_smiles
smiles = to_smiles_method(self, isomeric, explicit_hydrogens, mapped)
File "openff/toolkit/utils/openeye_wrapper.py", line 1323, in to_smiles
oemol = self.to_openeye(molecule)
File "openff/toolkit/utils/openeye_wrapper.py", line 1185, in to_openeye
"Programming error: OpenEye atom stereochemistry assumptions failed: "
Exception: Programming error: OpenEye atom stereochemistry assumptions failed: None != S
Diagnostics
I do not have a good solution for this. I record here my attempts to diagnose the problem.
One issue is that the RDKit and OEChem wrapper assign differ chiralities. Given the DrugBank_6182 from earlier,
rd_mol = rd_wrapper.from_file_obj(io.BytesIO(DrugBank_6182), "SDF")[0]
oe_mol = oe_wrapper.from_file_obj(io.BytesIO(DrugBank_6182), "SDF")[0]
def get_stereo_flags(mol):
return [(i, atom.stereochemistry) for i, atom in enumerate(mol.atoms)
if atom.stereochemistry is not None]
print(" RDKit:", get_stereo_flags(rd_mol))
print("OEChem:", get_stereo_flags(oe_mol))
prints:
RDKit: [(12, 'S'), (13, 'S'), (14, 'R'), (15, 'S'), (16, 'S')]
OEChem: [(13, 'S'), (14, 'R')]
The assumption is that the Molecule-defined stereochemistry will be accepted by OEChem, when it doesn't in this case where the Molecule was created by RDKit.
Tracking that down, those assignments are visible in the molblock:
-0.0593 0.0296 -1.7032 C 0 0 1 0 0 0 0 0 0 0 0 0
0.8494 1.2095 0.3359 C 0 0 1 0 0 0 0 0 0 0 0 0
0.6975 -1.3101 0.2998 C 0 0 2 0 0 0 0 0 0 0 0 0
1.5002 -0.1022 0.8274 C 0 0 1 0 0 0 0 0 0 0 0 0
-1.4137 0.0787 0.4257 C 0 0 2 0 0 0 0 0 0 0 0 0
...
13 43 1 6 0 0 0
14 16 1 1 0 0 0
...
However, manually changing those values does not seem to affect anything. I believe RDKit is doing all that perception itself. (If all values are set to 0 then RDKitToolkitWrapper complains about unspecified stereochemistry.)
CHEMBL241707
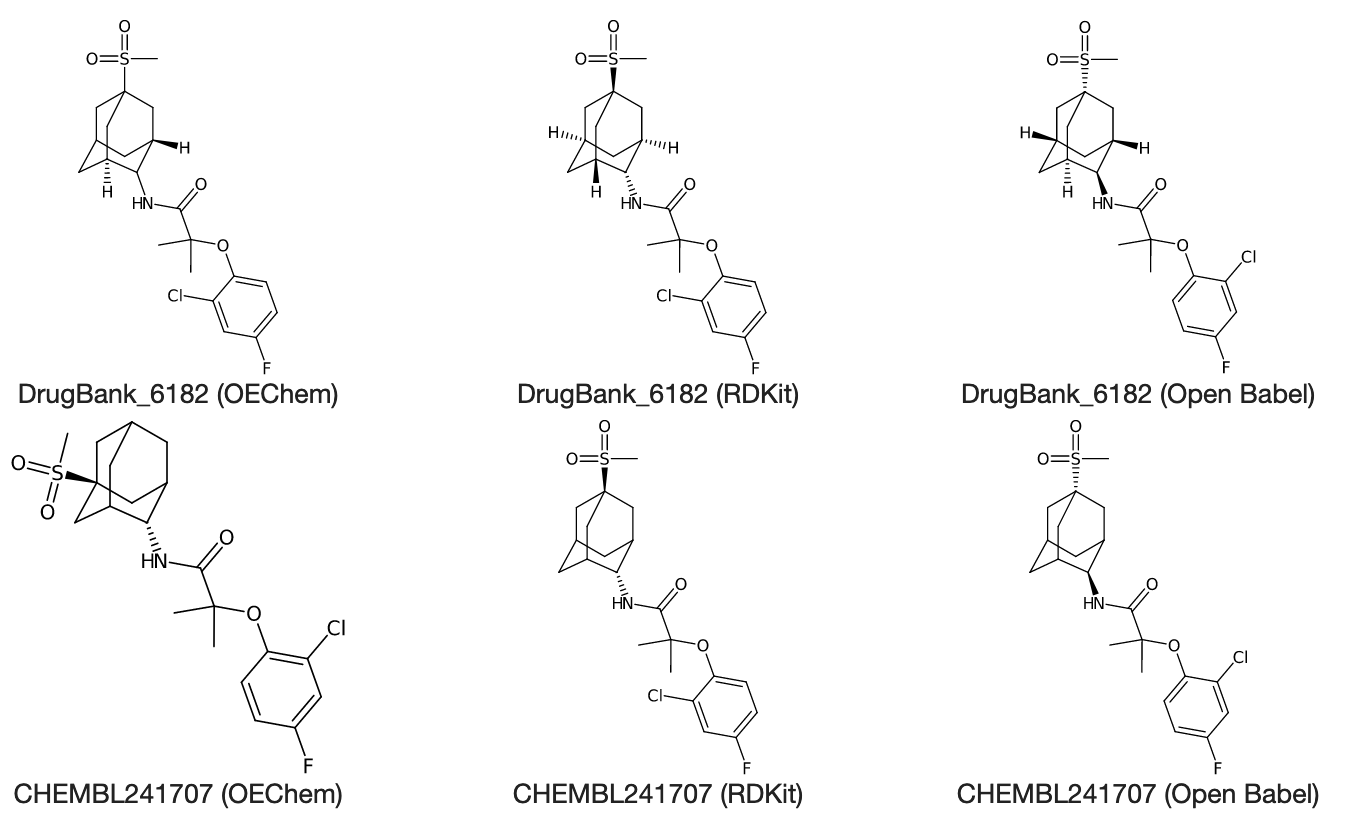
The DrugBank_6182 record is 100% similar to CHEMBL241707, though they don't have the same canonical SMILES.
CC(C)(C(=O)NC1[C@@H]2CC3C[C@H]1CC(C2)(C3)S(=O)(=O)C)Oc4ccc(cc4Cl)F DrugBank_6182 (OEChem)
CC(C)(Oc1ccc(F)cc1Cl)C(=O)N[C@H]1[C@H]2C[C@H]3C[C@@H]1C[C@](S(C)(=O)=O)(C3)C2 DrugBank_6182 (RDKit)
Fc1ccc(c(c1)Cl)OC(C(=O)N[C@@H]1[C@@H]2C[C@@H]3C[C@H]1C[C@](C2)(C3)S(=O)(=O)C)(C)C DrugBank_6182 (Open Babel)
CC(Oc1c(cc(cc1)F)Cl)(C(=O)N[C@H]2C3C[C@@]4(CC2CC(C3)C4)S(=O)(=O)C)C CHEMBL241707 (OEChem)
CC(C)(Oc1ccc(F)cc1Cl)C(=O)N[C@H]1C2CC3CC1C[C@](S(C)(=O)=O)(C3)C2 CHEMBL241707 (RDKit)
Fc1ccc(c(c1)Cl)OC(C(=O)N[C@@H]1C2CC3CC1C[C@](C2)(C3)S(=O)(=O)C)(C)C CHEMBL241707 (Open Babel)
Here's what those look like in CDK's depictor:
Trying CHEMBL241707
If I used CHEMBL241707.sdf as my input, I find the RDKitToolkitWrapper doesn't handle it:
openff.toolkit.utils.exceptions.UndefinedStereochemistryError: Unable to make OFFMol from RDMol: Unable to make OFFMol from RDMol: RDMol has unspecified stereochemistry. RDMol name: CHEMBL241707Undefined chiral centers are:
- Atom C (index 5)
- Atom C (index 7)
Disable assumption check
If I disable the assumption check then repr() works and rd_mol.is_isomorphic_with(rd_mol) is True.
oe_mol.is_isomorphic_with(rd_mol) is still False, but oe_mol.is_isomorphic_with(rd_mol, atom_stereochemistry_matching=False) is True.
Interesting! So the DrugBank file defines stereo on 5 atoms and 2 single bonds, and the ChEMBL file defines stereo on 0 atoms and 2 single bonds.
It looks like, when reading from the DrugBank file, RDKit stores definitions on 5 stereocenters (one for each non-CH2 atom in the adamantane), while OpenEye only stores it on two of them. I'm guessing that OpenEye has some heuristic for adamantane chirality that understands how the stereocenters are coupled, and can understand the chirality of all the atom stereocenters in our adamantane by just knowing the chirality of one or two.
What's strange is that the depiction of the OE molecule from the Drugbank file doesn't have any stereo assigned for the adamantane atom that connects to the NH. That would seem to be a significant stereocenter to me, and yet OE didn't raise an error. Is there something about the symmetry of the substituted adamantane that removes the need for that to be a stereocenter?
When reading from the ChEMBL file, where only two stereocenters are defined, RDKit complains about missing stereo info for two others. So I take this to mean that RDKit requires stereo info for many centers (apparently 4), and is happy to store stereo info for more (because in the DrugBank molecule, it stores stereo info for 5 atoms).
Severity
This seems to be two problems: 1) RDKit and OpenEye ToolkitWrappers can't hand this molecule back and forth to each other, and 2) the RDKit ToolkitWrapper expects more stereo information than the ChEMBL version of this molecule contains.
For 1) it's bad that the two toolkits get a different graph molecule from the same MOL block. That said, it seems like both toolkits are fully internally consistent - that is, if RDKit can read the MOL block, it can generate conformers and parameterize the molecule - so the only problem is when both backends are handing the molecule back and forth. I think there are relatively few situations where the user would have access to OpenEye, but manually override and read the MOL block with RDKit instead, and then try to do molecule operations with OpenEye. So I think this would be "low severity".
For 2), this could be due either to the ChEMBL molecule being underdefined, or RDKitToolkitWrapper thinking it needs more stereo info than it really does. I don't know which of these two is the case. The particular molecule in this issue is quite complex, but we may be able to make a minimal case of the problem with just an adamantane with two simple substituents, and then we could ask a real chemist how many stereocenters it has. Until we know how many stereocenters this molecule "really" has, I don't think we can propose a fix. Though I think the general statement of "the RDKitToolkitWrapper can't read a molecule straight from ChEMBL" makes this medium-severity until we dig deeper into it.
Proposed resolutions
If my above assumptions are correct, then we have a few ways we could resolve this:
- See if there's a way to "unpack" or "dumb down" the OpenEye adamantane stereochemistry so that each non-CH2 stereocenter is defined, even if OETK thinks this is redundant.
- Put OE's adamantane stereochemistry heuristics into RDKit, which would be a nice feature contribution, but may be an unwelcome change to its core stereo model, and would require a ton of work.
- Ask a "real chemist" how many stereocenters the simple dual-substituted adamantane described above has.
I processed 45,043 records from ChEBI and did not find additional cases where OpeneEye atom stereochemistry assumptions failed.
I found several records where bond stereochemistry failed. See https://github.com/openforcefield/openff-toolkit/issues/1011 .
(Again, these are using the path RDKit -> RDKitToolkitWrapper -> Molecule -> OpenEyeToolkitWrapper -> OpenEye, which is not likely to be a common use case.)