[FEATURE REQUEST] anvi-interactive BLASTP for amino acid fastas
The need
The BLAST functionality of the interactive interface can only use nucleotide sequences. In the case of working with amino acid sequences in the interface provided by the --fasta-file flag, it would be really nice to be able to blast the amino acid sequence.
The solution
The interactive interface could be able to tell what kind of fasta is being provide by the --fasta-file flag then adjust which BLAST option are available for the user.
Beneficiaries
This help users who want to BLASTP sequences from an amino acid phylogenetic tree.
Thanks! Here is a reproducible way to set up a test case with amino acid sequences:
cd INFANT-GUT-TUTORIAL
# extract Ribosomal_L1 seqs
anvi-get-sequences-for-hmm-hits -c CONTIGS.db \
-p PROFILE.db \
-o seqs-for-phylogenomics.fa \
--hmm-source Bacteria_71 \
-C default \
--gene-names Ribosomal_L1 \
--concatenate-genes \
--return-best-hit \
--get-aa-sequences
# Calculate tree
anvi-gen-phylogenomic-tree -f seqs-for-phylogenomics.fa \
-o phylogenomic-tree.txt
# make fasta with simple headers and no gaps
sed 's|\(^.*\) .*|\1|' seqs-for-phylogenomics.fa > seqs-for-phylogenomics-renamed.fa
seqkit seq -g seqs-for-phylogenomics-renamed.fa > seqs-for-phylogenomics-renamed_no_gaps.fa
# visualize
anvi-interactive --tree phylogenomic-tree.txt \
-p temp-profile.db \
--title "Pylogenomics of IGD Bins" \
--manual --fasta-file seqs-for-phylogenomics-renamed_no_gaps.fa
and here's seqs-for-phylogenomics-renamed_no_gaps.fa
$ cat seqs-for-phylogenomics-renamed_no_gaps.fa
>E_facealis
MAKKSKKMQEALKKVDATKAYSVEEAVALAKDTNIAKFDATVEVAYKLNVDPKKADQQIR
GAVVLPNGTGKTQTVLVFAKGEKAKEAEAAGADFVGDDDMVAKIQGGWFDFDVVVATPDM
MATVGKLGRVLGPKGLMPNPKTGTVTMDVTKAVEEVKAGKVTYRVDKAGNIHVPIGKVSF
DNEKLVENFNTINDVLLKAKPSTAKGQYIKNISVTTTFGPGIHVDQASF
>S_epidermidis
MAKKGKKYQEAASKVDRTQYYSVEEAIKLAKETSVANFDASVEVAFRLGIDTRKNDQQIR
GAVVLPHGTGKSQRVLVFAKGDKITEAEEAGADYVGEADYVQKIQQGWFDFDVVVATPDM
MGEVGKLGRVLGPKGLMPNPKTGTVTMDVKKAVEEIKAGKVEYRAEKAGIVHASIGKVSF
DEEKLVDNFRTLQDVLAKAKPASAKGTYFKSVAVTTTMGPGVKVDTSSFKL
>L_citreum
MTQKHGKKYTAALAKVEAEKNYALNDAVALVKEIDYANFDASVEVAFKLNVDTRQADQQL
RGAVVLPNGTGKDKKVIVFAQGDKAKEAEAAGADVVGAADLVAQIQGGWMDFDTAIATPD
MMAQVGRVARILGPKGMMPNPKTGTVTMDVAKAVSDAKGGQVTYRTDRDGNVAVPVGRVS
FEEGKLAENIKTIADTVLKARPAAVKGTYVQHVSIASTFGPAVALDLNTL
>S_aureus
MAKKGKKYQEAASKVDRTQHYSVEEAIKLAKETSIANFDASVEVAFRLGIDTRKNDQQIR
GAVVLPNGTGKSQSVLVFAKGDKIAEAEAAGADYVGEAEYVQKIQQGWFDFDVVVATPDM
MGEVGKLGRVLGPKGLMPNPKTGTVTMDVKKAVEEIKAGKVEYRAEKAGIVHASIGKVSF
TDEQLIENFNTLQDVLAKAKPSSAKGTYFKSVAVTTTMGPGVKIDTASFK
>P_rhinitidis
MPKRGKKYQDSVKLIDKSNLYDVNEALDLVTKTAKANFDETVELAVRLGVDPRHADQQVR
GTVVLPNGTGKEVKVLVLAKGEKIKEAEAAGADYAGGEEYVEKIQNENWFDFDVLIATPD
MMGVVGKIGRVLGPKGLMPNPKSGTVTFDIEQAVKETKAGKVEYRVDKAAIINVPIGKVS
FGVDKLAENFKVIADAIIKAKPAAAKGRYLKSVTVSSTMGPGVKVNGSKLMEK
>C_albicans
METVELQVGLKNYDPQRDKRFSGTLKLPQVPRPNMTICIFGDAFDVDRAKSLGVDAMSVD
DLKKLNKNKKLIKKLAKKYNAFIASEVLIKQIPRLLGPTLSKAGKFPTPVSHNDDLYSKV
TDVKSTIKFQLKKVLCLAVAVGNVDMEEDVLVNQIMMAANFLVSLLKKNWQNVGSLVIKS
TMGPSFRIY
>P_avidum
MKRSKAYRAAAEKVNFDQLYSPEEGLALVASGASAKFDETVDVAIRLSVDPKKADQMVRG
TVNLPNGTGKTARVLVFATGEKAEAARAAGADEVGDDDLIAKVQGGYLDFDSVVATPDMM
GKVGRLGRVLGPRGLMPNPKTGTVTMDVAKAVSDIKGGKIEFRTDRYANLHFLIGKVSFG
PEKLVENYYAALDEILRLKPNSAKGRYLRKVTVSSTMGPGVQIDPASARVAE
@mschecht what's the database that blastp calls? nr/refseq, etc
@meren it looks like we need to modify the get_sequence_and_blast function in the data>interactive>js>utils.js file to query the backend for the AA sequence in question. Or we can return the AA sequence in addition to the nucleotide sequence in the same existing backend call.
I am not sure about it, @matthewlawrenceklein.
This is the FASTA file provided by the user to the interface.
The code assumes that FASTA file always contains DNA sequences, but clearly it can also be AA sequences. IF the FASTA file contains AA sequences, we should target different databases on NCBI for search.
I think the best way to solve this is to figure out if a sequence is DNA or AA right before forming the BLAST call. For instance, the ratio of the nucleotides A + T + C + G in a given sequence makes more than 75% of all nucleotides in it, we can assume it is DNA. If not, we can say it is AA, and perform the search based on that. I don't think this requires any modification in the backend.
@meren to clarify, AA/DNA sequences aren't explicitly passed to the frontend as such, so it should therefore be a process on the frontend to determine whether or not a given sequence is DNA or AA? Would this be some new novel introspection, or does this occur elsewhere in the interface?
OK. I went back to the code, and realized that what I'm proposing is not going to work :/ Because we're trying to change the items in this menu:
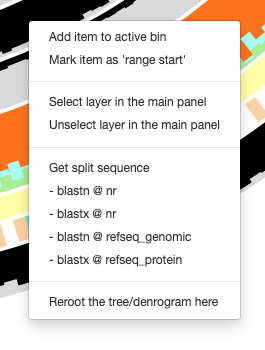
If we have AA sequences, we shouldn't show in this menu blastn, but blastp, and so on. Which means the backend needs to tell the interface if sequences are DNA or AA sequences :/ Which I added now in #1902.
But the frontend is designed so poorly, I am not sure how to solve this there since menu items are added before we know the nature of sequence for a given item. One way to solve this is to remove 'program' names and only keep databases, and decide program names here:
function get_sequence_and_blast(item_name, program, database, target) {
$.ajax({
type: 'GET',
cache: false,
// async: false is important here. DO NOT REMOVE.
// If direct call chain between event handler and the code that opens new window is broken
// chrome popup blocker will not allow opening new window.
// async: false does not use asynchronus callbacks so protects direct call chain.
async: false,
url: '/data/' + target + '/' + item_name,
success: function(data) {
if ('error' in data){
toastr.error(data['error'], "", { 'timeOut': '0', 'extendedTimeOut': '0' });
} else {
var sequence = '>' + data['header'] + '\n' + data['sequence'];
fire_up_ncbi_blast(sequence, program, database, target)
}
}
});
}
I.e., if data has sequence_type and it is DNA, we use blastx, blastn etc. If AA, we use blastp, etc.
It is a thorny one. In an ideal world we would redesign this, but no one has time for that. So I am a bit discouraged to implement this feature request :(