[BUG] [SOLVED] anvi-run-kegg-kofams HMM annotation fails and subsequently hangs
Solution
The problem is due to a bug in HMMER v3.3.1. If you downgrade it the following way, the error will be resolved:
conda install -c bioconda hmmer=3.2.1
Short description of the problem
both anvi-run-hmms and and anvi-run-kegg-kofams encounter errors when running hmms on specific databases, but not the same one. The errors cause the programs to hang.
anvi'o version
Anvi'o .......................................: hope (v7)
Profile database .............................: 35
Contigs database .............................: 20
Pan database .................................: 14
Genome data storage ..........................: 7
Auxiliary data storage .......................: 2
Structure database ...........................: 2
Metabolic modules database ...................: 2
tRNA-seq database ............................: 1
System info
Linux, RedHat 8.3. Installed v7 following updated install instructions (not the yml file)
Detailed description of the issue
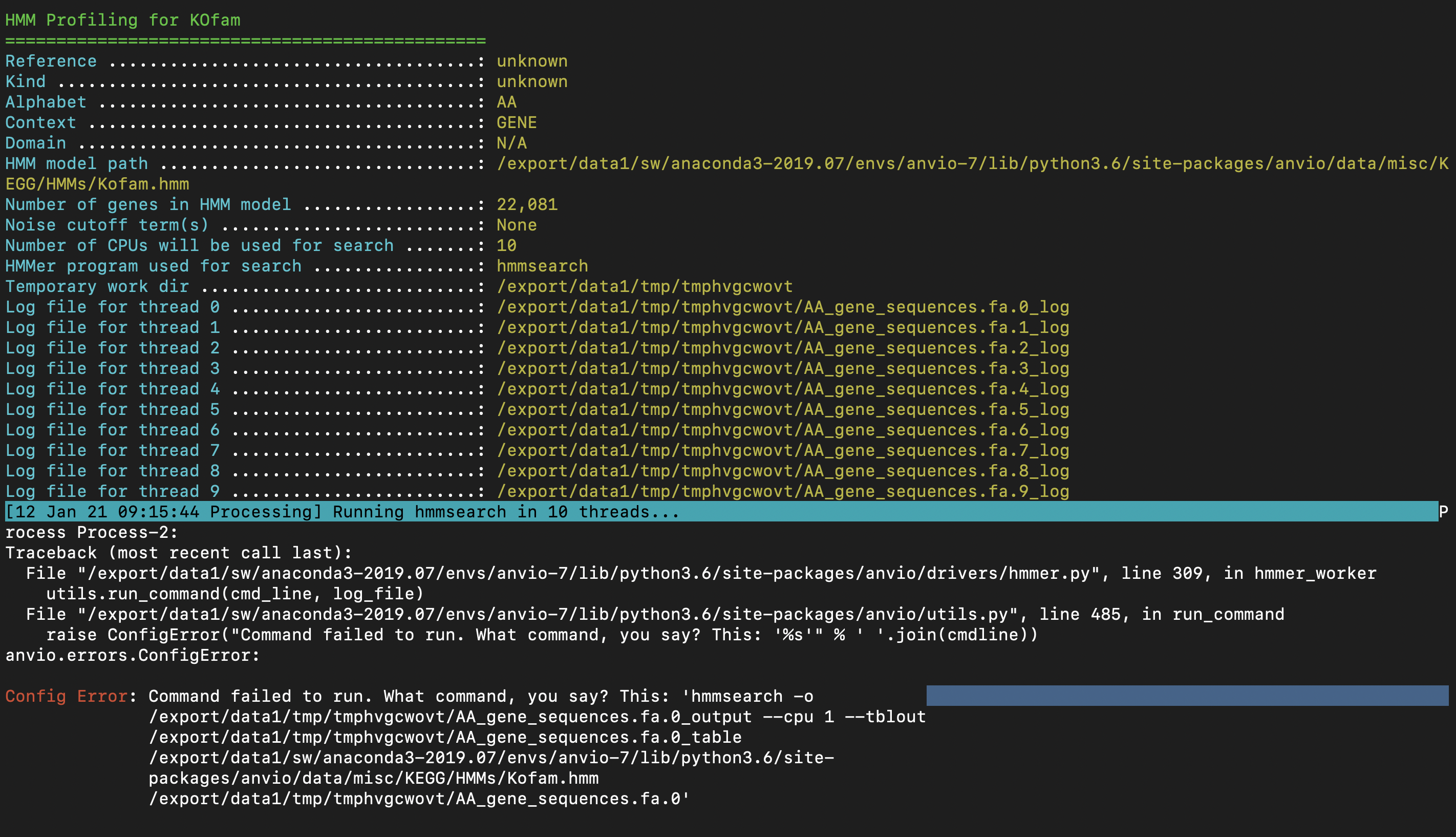
When running hmms through two anvio commands, the run occasionally throws an error and subsequently hangs. This is a database specific issue, as I have only encountered it on specific databases. In the case detailed in the screenshots below, anvi-run-kegg-kofams ran fine on 300+ dbs before hitting a snag on this specific one.
The other instance of the problem, encountered using anvi-run-hmms is detailed in two screenshots at the bottom of this issue

Files to reproduce
for anvi-run-kegg-kofams, it happens on a contigs db created from GCA_003647425. I'm happy to share the specific db if needed. anvi-run-hmms runs fine on this specific db
For anvi-run-hmms I don't know on which db it happens, because the log output does not seem to store the db name anywhere when run as part of a for loop (will create specific issue for that).
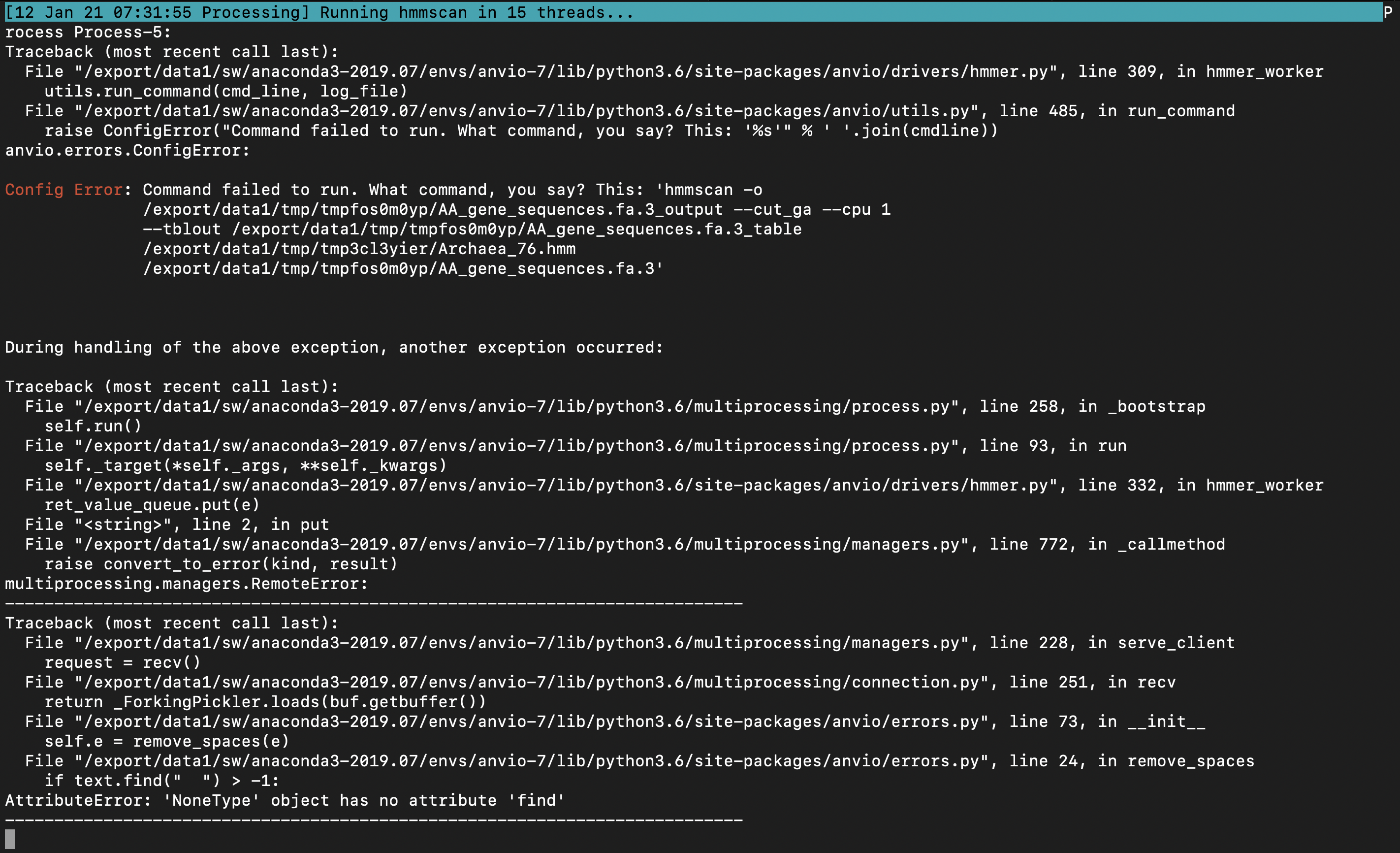
anvi-run-hmms issue:

I'm sorry you run into these annoying errors, @dspeth.
I will start by addressing the fact that anvi-run-hmm does not log which db it is working on. Since you are not using the development branch, I will suggest you to edit a particular file as I will make minimal changes in the code.
Hi @meren, ah no worries about the logging. i won't create a feature request then, and patiently await 7.1 :P To your credit, I had noticed the lack of db identifying output before, but just never bothered me because everything always ran smooth :D
Hey @dspeth,
If you want to do some hacking, feel free to run this on your terminal, which will tell you a file path:
python -c "import anvio.tables.hmmhits as h; print(h.__file__)"
Open it in your text editor, and either jump to 146, or search for ("HMM sources in the file (which occurs only once). Once you find the line, add the following line above that line:
self.run.info("Contigs DB", self.db_path)
So it looks like this:

If you re-run your HMMs, then, you should see the DB path (coding for anvi'o is this easy actually, and we just pretend that we're doing a lot of work (please keep this hack between us hehe)).
It would be helpful to get that contigs db with anvi-run-hmms error so we can address this unique error we never run into :)
I'll get you the contigs db in about 5hrs @meren
I keep a local copy of the gtdb species representatives as anvio dbs for easy use in phylogenomics, pangenomics etc. Was attempting to run hmms on them after the port from 6 to 7 because I wanted to update the rRNA hmms. So it's one of the 30000+ dbs :D
tangentially related: does anvi-run-hmms support a list of hmms? I tried a few options to just run the rRNA ones, but didn't get it going and decided to brute force it instead, and rerun all hmms on all dbs because it was fewer commands.
on another note, did you need the specific contigs db for the anvi-kegg-kofam hmm issue?
Wait. I think I can solve the anvi-run-hmms issue without needing a database.
Don't worry about it! It has nothing to do with HMMs I realized, so don't start everything over :)
I keep a local copy of the gtdb species representatives as anvio dbs for easy use in phylogenomics, pangenomics etc. Was attempting to run hmms on them after the port from 6 to 7 because I wanted to update the rRNA hmms. So it's one of the 30000+ dbs :D
We are actually planning to create a continuously integrated archive of these genomes online. You are beating us to it :)
tangentially related: does anvi-run-hmms support a list of hmms? I tried a few options to just run the rRNA ones, but didn't get it going and decided to brute force it instead, and rerun all hmms on all dbs because it was fewer commands
So you are asking for -I command to accept a comma-spearated list of HMM sources? We can do that!
hold up, forgot to add something. While the error seems the same in both anvi-run-hmms and anvi-run-kegg-kofams, the handling of the progress after the keyboard interrupt is different.
anvi-run-kegg-kofams, being nice (a little too nice) deletes all the temp files.
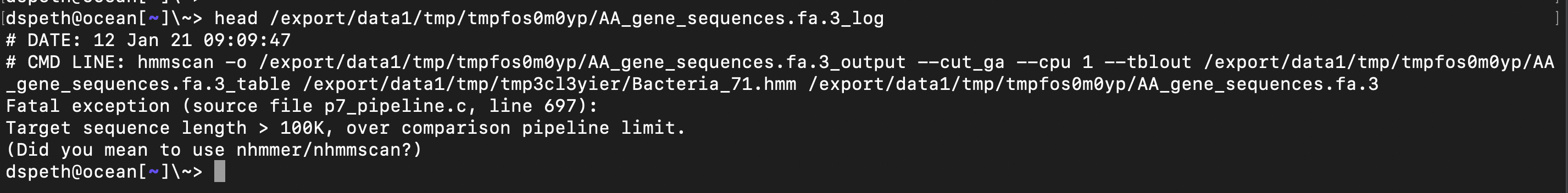
anvi-run-hmms, the wild child, just says f*** it and moves on, leaving the temp files available. This is what the log says:
and on the -I flag:
yes, that was exactly what I was asking. That way, it becomes easier to update the "Ribosomal RNAs" hmm to the new ones with 6 distinct hmms
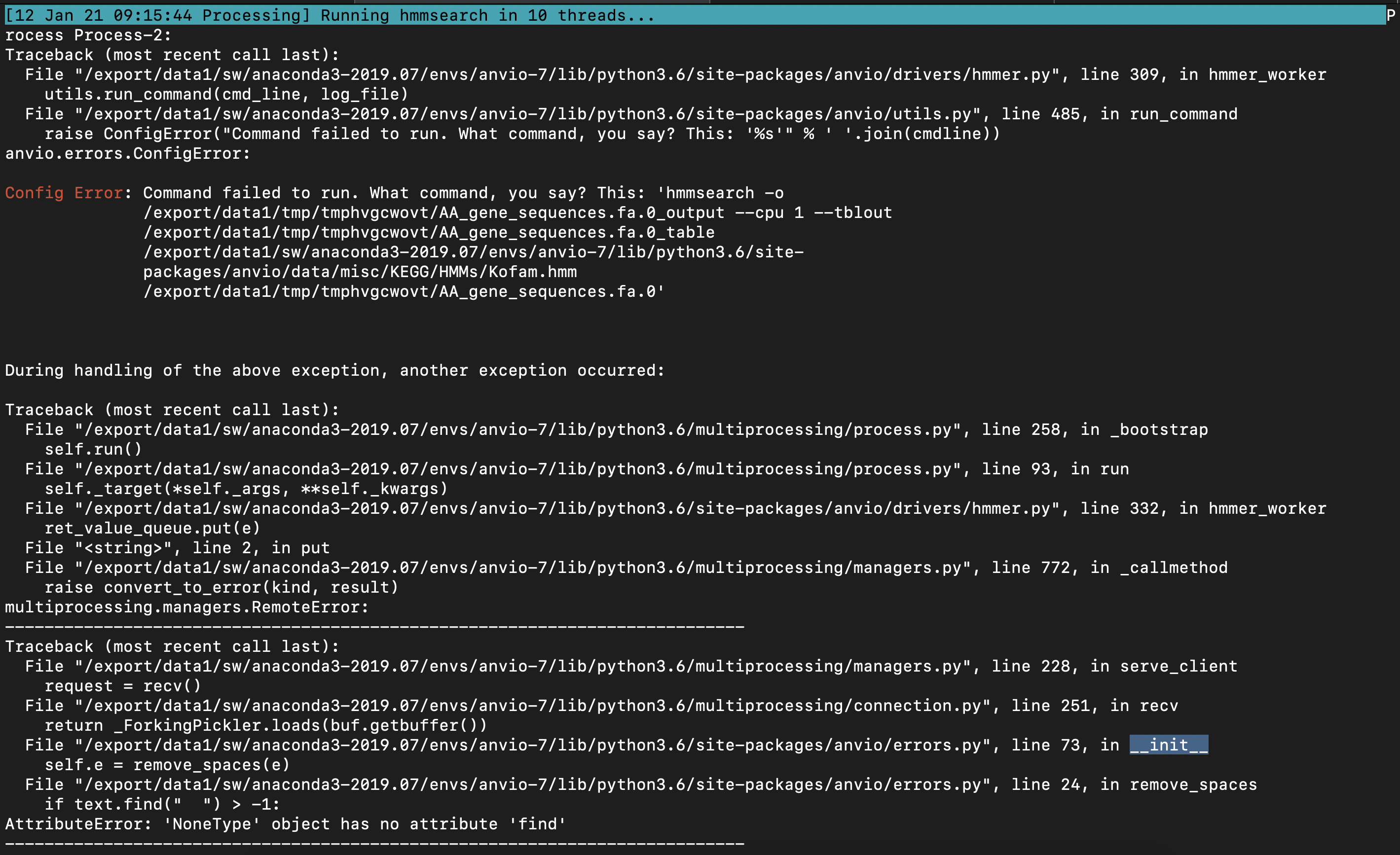
e2d861145e4fb72713d60f857fa5ef027822f31e solves the cryptic anvi-run-hmms error due to "'NoneType' object has no attirbute 'find'", but clearly there is something even deeper here.
If I'm reading your message correctly, You should find in that temp dir the AA_gene_sequences.fa.3 with a gene that is longer than 100K amino acid residues :/
Sounds like a Nobel to me :)
Sounds like a Nobel to me :)
Although I am not sure if it should be given to GTDB, Prodigal, or Daan at this point :)
If you still have access to AA_gene_sequences.fa.3, can you confirm if there is a weird looking gene in it?
I had looked, and there are genes with many many X residues in it, just didn't think it was over 100K. But now I'm second guessing myself and checked properly, and indeed it is longer than 100K residues. Here's the (shortened fasta):
>730 MPGGRWARPCSRSTCTSRAAWARAKCRSFSPAAPCPRPPTAGGRASGPGWAAGAARRADXXXXXXXXXXXXXXXXXXXXX XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX 1298 lines of X residues XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXQGARECLRH GSARHAVLRRTADGAVIITGEISEPQAHLARETGVAFLAAGHHATERYGAPAAAAHVAAALGIEHRFIEIDNPA
Now I'm curious if this is a genome with a crazy number of Ns, or is it anvi'o screwing things up somehow :/
Still, doesn't seem to solve the anvi-run-kegg-kofams issue. Possibly unrelated. Let me see if I can run this with --debug to preserve temp files
I wouldn't do that for that many genomes, @dspeth :( Let's ask @ivagljiva for the best practice.
Perhaps we should generate a contigs db for GCA_003647425.
as for the other one, I'll tell you later today :D but since it is a gtdb genome that shoudn't be the case. Otherwise I can go complain with donovan and phil
ah, @meren the failure for anvi-run-kegg-kofams was on an unrelated project, with only 427 genomes in it, and there I do know the culprit db specifically.
Ironically, anvi-run-hmms runs smoothly on it
here's the --debug output from the moment anvi-run-kegg-kofams throws an error.
Nothing quite as obvious as the problem with anvi-run-hmms seems to be happening, but I might be checking the wrong log files?
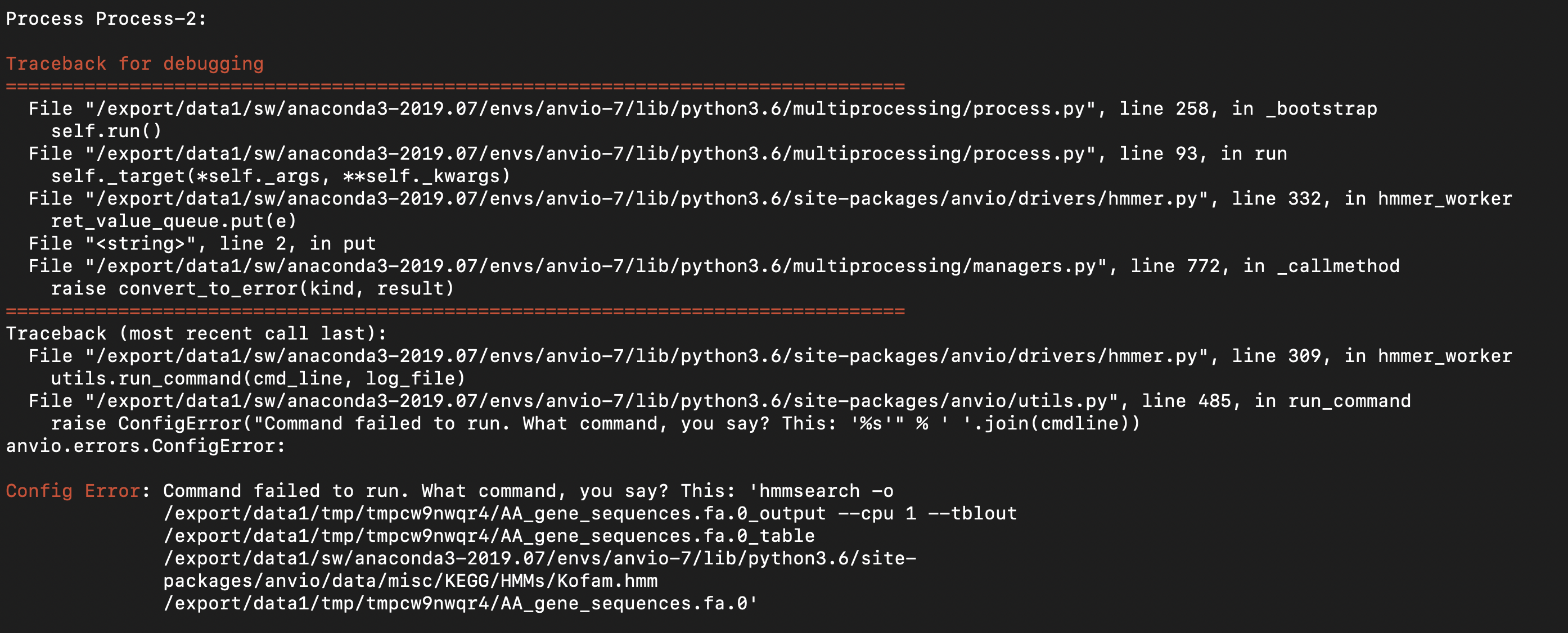
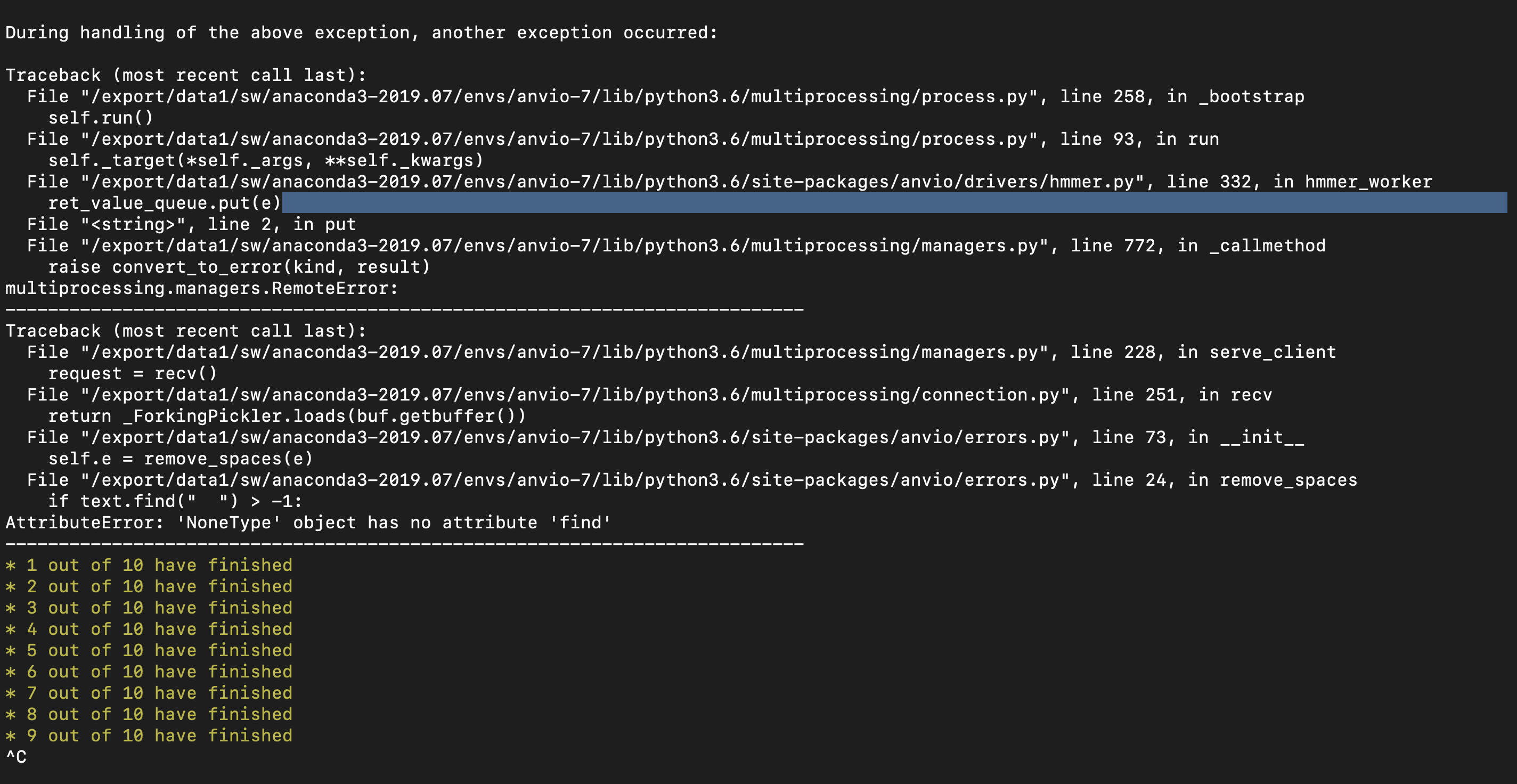


How long does it take for you to get this error on that other project, @dspeth? Are you able to share the contigs db with @ivagljiva privately so she can take a look and isolate the error?
@dspeth, #1639 now solves this tangential point in the development branch and will be in v7.1 unless you change your mind and switch to the development branch :)
tangentially related: does anvi-run-hmms support a list of hmms? I tried a few options to just run the rRNA ones, but didn't get it going and decided to brute force it instead, and rerun all hmms on all dbs because it was fewer commands
So you are asking for -I command to accept a comma-spearated list of HMM sources? We can do that!
@meren, thanks!
I emailed iva the contigs db. As I mentioned buried somewhere above,. I generated this particular db from a public mag (GCA_003647425), but in a previous version of anvio. I'm guessing 6, but could even be 5.
@meren the normal runtime for anvi-run-kegg-kofams is ballpark 2 minutes with these settings on our server, so i run into this issue with this particular db before that time. when running single threaded, the same happens
Thank you very much for your help, @dspeth. The version of the contigs db should not affect anything. I'm sure we will discover a bug with our current codebase and fix it :)
@dspeth, I managed to replicate your error on my system. Yay :)
It seems like only thread 0 is failing. So I took the portion of the sequences that thread 0 was working on, and ran the same hmmsearch command that it was running on that file (AA_gene_sequences.fa.0), outside of anvi'o. This is what I got:

A segfault is coming from hmmsearch on this chunk of sequences. :/ This segfault did not happen when I tried the same strategy with the second chunk of sequences (AA_gene_sequences.fa.1), so for whatever reason it is input specific.
And it seems like our exception-catching does not quite know how to handle this segfault, so it hangs. I am not sure how to fix it just yet.
And meanwhile, there was this just posted on Slack with the same error, so this is far from being an isolated issue.
There must be something weird with the sequences, and we probably want to be able to catch that way earlier, perhaps all the way at the anvi-gen-contigs-database stage.
If it is not too big, can you please make this contigs db available in this issue?
As I was suspecting, the contigs.db contains multiple genes over 10K nucleotides. I wonder if the new version of HMMER has a problem with that :/
sqlite3 -header GB505_GCA_003647425.db \
'select gene_callers_id, stop - start from genes_in_contigs where stop - start > 5000;'
| gene_callers_id | stop - start |
|---|---|
| 29 | 11448 |
| 39 | 7416 |
| 95 | 7761 |
| 224 | 5490 |
| 368 | 10230 |
| 483 | 9912 |
| 723 | 7011 |
| 783 | 5556 |
| 868 | 7194 |
| 870 | 6165 |
| 969 | 5631 |
| 1148 | 5448 |
| 1245 | 5160 |
| 1542 | 5571 |
| 1555 | 5811 |
| 1693 | 5553 |
| 1814 | 6087 |
| 2095 | 5607 |
| 2352 | 5250 |
| 2457 | 5082 |
Here is what I did:
# get the contigs:
anvi-export-contigs -c GB505_GCA_003647425.db -o contigs.fa
# get the gene calls
anvi-export-gene-calls -c GB505_GCA_003647425.db -o gene-calls.txt --gene-caller prodigal
# create an external gene calls only with gene_caller_id 29 (11K nts):
head -n 1 gene-calls.txt > new-gene-calls.txt
grep '^29 ' gene-calls.txt >> new-gene-calls.txt
# generate a new contigs database with it:
anvi-gen-contigs-database -f contigs.fa --external-gene-calls new-gene-calls.txt -o contigs.db
External gene calls parsed fine:
EXTERNAL GENE CALLS PARSER REPORT
===============================================
Num gene calls in file .......................: 1
Non-coding gene calls ........................: 0
Partial gene calls ...........................: 1
Num amino acid sequences provided ............: 1
- For complete gene calls ..................: 0
- For partial gene calls ...................: 1
Frames predicted .............................: 0
- For complete gene calls ..................: 0
- For partial gene calls ...................: 0
Gene calls marked as NONCODING ...............: 0
- For complete gene calls ..................: 0
- For partial gene calls ...................: 0
Gene calls with internal stops ...............: 0
- For complete gene calls ..................: 0
- For partial gene calls ...................: 0
This run with no problem, which uses hmmscan:
anvi-run-hmms -c contigs.db
This gave me the following error, which uses hmmsearch:
anvi-run-kegg-kofams -c contigs.db
WARNING
===============================================
An exception was thrown in one of the worker threads (see output below for
details).
✖ anvi-run-kegg-kofams encountered an error after 0:00:09.178717
Config Error: Command failed to run. What command, you say? This: 'hmmsearch -o /var/folders/g
w/5mdblzs94gsb1ss44llgl3_h0000gn/T/tmpkldgr9mi/AA_gene_sequences.fa.0_output
--cpu 1 --tblout /var/folders/gw/5mdblzs94gsb1ss44llgl3_h0000gn/T/tmpkldgr9mi/AA
_gene_sequences.fa.0_table
/Users/meren/github/anvio/anvio/data/misc/KEGG/HMMs/Kofam.hmm /var/folders/gw/5m
dblzs94gsb1ss44llgl3_h0000gn/T/tmpkldgr9mi/AA_gene_sequences.fa.0'
Indeed, this is the gene sequence:
>29
QVAIGGQSIIQIPMDEESTIWELNHGLGVLPVDVICIDGFNSPIEPDRVEYLNSNFIRLTFDSTSQWGTCLCISGGGQTG
ATGDSGASGGTGDSGGTGATGQVAIGGQSLTEIPESQESATWTIHHGLGVIPVNFVCIDNSQILLHPDSVTYVDLNTIEF
EFEFEQWGRCLCISGGGMTGNTGHSGGTGGLGGSGGTGATGQVAIGGQSVTEIDLIDESATWNIEHGLGIIPVLVICVDE
NDDIIVPDEIEYLSGNAVELTFDVGHHGKCLCISGGGQTGATGASGGSGGTGDSGGTGSTGQVAIGGQSTTIIAEDENST
IWTIHHALGTDLVQTICVDENNDIIIPSNINLVSLNSVEFTFDDPHWGRCLCISGGGMTGNTGASGGTGGLGGSGGTGAT
GQVAIGGQSLTTVPASEISAIWNIHHGLGSMPVEVICIDNTGLKLYPENIQYSDANNLTVTFSGSEFGRCLCISGGGMTG
STGHTGASGGTGNTGNTGATGQVAIGGQSITDVASTDTSSVWEIHHGLGIIPVHVICMDEAYQSIEIDKVIFLSTNSIQV
ELPAPHYGKCLCISGGGMTGDTGDTGGSGGLGETGGSGATGQVAIGGQSLNTIESTETSSTWEIHHGLGVMPVFVSCLDH
NNIVITPSDVSYINHNVIQLTFDVSYYGTCMCISGGGMTGSTGHTGHTGGIGGSGGTGSTGQVAIGGQSMTNIPQTEESA
VWFVTHGLGSKFVSIHCFDHMDEIIIPTDVVFSGMNNIELTFDGSKYGTCMCISGGGMTGATGHTGHTGGIGGSGGTGAT
GQVAIGGQSISEIPISEESSDWNIHHGLGISPVQIICIDQNDELIYPSAIDFITINSAMVTFSSPKHGKCLCISGGGMTG
DTGDTGGSGITGGTGMTGGLAIGGQYNEVIGFDDKDDTWEIVHGLNCMPVAVVCVDQNDDLLYPENIEFSTGNSVTVQFT
PGSELYGRCLCVSGGGITGGTGHTGGSGGTGDTGHSGGTGQIAIAGNVIFNISEIDVIDNNDDTLWEWHIRHHLESYPVI
ECIDIEGNSIQIHHIKYLPNKEDVIVQFSPNDIDEAPIGKCICVSGGGQTGERGNFQIDQAYEEFNDVDVDNIRQVVIEN
ELTYYLISIIEDVRVHSLLPGISTPDMSRHMIMYDGSLWHDYGEYIGRTGHTGGSGGTGERGDAVIIGQSVIEFEFPDQT
WVVDHNLGIQFVAFQCFDANGNALPYATARYIDDTTLRINFESPQIGHCLCTSGGGLMGKPGETGGTGDTGEPGDAGPVG
RDGRDGIQGDPGEVGPAGVLGDAGPPGCPGAIGPIGRDGRDGEKGDLGPAGPMGFPGAIGPAGPLGTSGESGATGSKGDP
GIGAMTMVPIDSPATSWVVNHNMGVQYMDIVCVDYSDQMIPFESVNFTDINNLTVTFDTEKYGKCFCITGGGATGATGGP
GKDATGGVLDWEENTGPIWEIEHNLDSTSVIVLARNFDGTIIYPTNIIYVDRNHIDLHFGESQSGFATISSGGGASGGSG
GSGTTGATGATGDKGGMGDTGNTGNTGPTGGSGGIGISGGTGKAGIGSLETYSQLVSDTTWDFNHGFNTTELAVQCYDEF
NNYLVPFNVIFTTTMVTVTFNSAAKGRIIIISGGGETGGTGISGNVGETGARGDLGPRGYDGLPGVKGERGETGADGKGT
LQEYFVSVADDTWIISHTLNTNMLAVQCYDEADNLLDPVNVQYPNDNEIIITFPSDKTGRVVVISGGGETGGSGGIGDTG
GSGKTGDIGSVGPRGYDGNPGQPGIDGIDGDKGDDGNSGGTGNSGSTGGAAKGNLYTYTQNTPSDSWVIIHPLGTQTIAV
QCYDLSNQLFHHKSVTIIDENTLSINFDPAATGKAILISGGGQTGGTGNIGSRGYDGDPGADGIPGDGGETGGTGAEGEQ
GDTGGTGHFEFLHTATGGSGGSGETGGSGGSGTTGGSGETGGSGHFGQPFNINEVFNEWTDLNRVEVENGGHITYEEPYY
VIMVVNDMRSEPYLPNEINVNVNRHCLAWDGTYWRDWGPIVGPTGGSGSSGRMGIDGKPFKITEYFEEFTDATIPYIESN
YSDATNEDYYTFSIQFDVRYSKSATPGIVGDMSRHVIIYKGEFQDWGLIVGQSGGTGTTGGSGETGDTGGSGGSGGSGGS
GGSGGSGGSGGSGGSGGSGDSGGSGGSGGSGDIGLTGGTGSSTGESGGTGNSGGSGDTGADGSAVFSGGTGGSGGTGRQG
RNWSITDVENSFEDSDVDKYINRGTATSLDPYFIYIIDDIRTNNNQPPGMTYDVSGHCLMYNGSEWEDWGKLQGKTGGSG
GSGGSGGSGGSGGSGGSGETGDSGGTGHTGHSGIDGDTGGTGHTGSGVTIQSHYINFYDDDVISIESIHTSRYAISIQND
LRTNRSIPVGLPSDLSLHAIAYEGIGTDWSDWGPFVGPKGDTGTPGGSGGSGGTGIDGLFAGTGGTGTTGSEGPPAEPIT
IDEYFESFGETEVAYIIGAYPDATLENPYHFSVLSDDRTNQSSPVGLENNVSRHVILYTGSFIDWGLLIGDTGGSGGSGG
SGGTGYTGGSGGSGGSGGSGGSGHTGHTGKSGDTGDTGGSGGSGGSGGVGPIGKSFGVNEHIPLFTSTDISRIEADQFGV
ANELDPYVITILYDIRSNKNIPASIIGDKSEHALAYDGETWIDWGKLVGSTGGSGGSGHTGDRGESFRMDAFYPDFDETI
IAEVEALPGISTADVYSLSCFNDTRLNQSIPEGIAGDMSRRIMMWDGTSWYDWGIFIGLTGGSGGSGETGDSGGSGYTGG
SGGTGDSGGSGGSGGSGGSGGSGGSGGSGGSGGSGGSGGTGHRGQDFRVDQFYLNIYDADIADIEANSGGTIDDVYLVAI
QNDLRITKDGTVGPAGDVDRHAIVWDGVQWEDWGITVGETGGSGGSGGSGGSGGSGGTGDSGGSGGTGRQIIGGSAIYHQ
SISSNEWHVHHSLGFKQVVVECSNDSSEVVIPNSIVYTDDQNLIIYFSENTSGYAVCVSGGGMTGGTGSDTGSTGGTGTH
GVSGVIGIDGETGGTGSIGYTGGTSGTGGSGSIGITGGTGSAVSVGSKIYHQTVASDTWTITHNLLNRYVNVACFDSLDE
VFEPDSISIIDDNTIQILLSVAITGHAICVSGGGQTGGTGSDTGSTGGTGQTGDVGYVGEDGETGGTGATSGTGVSGGSG
GTGATGSASIVGATYHVQSVLSDTWNINHGLGQRYVIVECYDDNNEVIDVDSIIAVDANNIQILLTEAISGYATCISGGG
FTGGTGSDTGSTGGTGSTGDVGESGGTGGIGGTSGTGESGGSGSSGGTGNSVLAGAYIYTQSIASDVWNITHTLGQNHVI
VECYTSIGNVIIPQSITTLDANNIRIVFSSDIAGTAVLVSGGGMTGGTGSATGETGGSGGSGGSGGSGGTGMTSGTGATG
GSGGSGGTGEVALGGTYTHNQDVTNSTWNVIHNLKTQFVTVQCFDDSYGLVYPDEVILDDDNNLTVLFSESLTGYCVCIS
GGGQTGGTGSSTGESGGTGETGYSGGSGATGEGISGGSGGTGIQGIPGTAVATGGTGHTGNKGKFEIDETISELFNITID
EIEIRAIDSTSDIYVVNVMHDLRLDNQIPPGLEGDLSAHLIMYDTTSWVDWGNFRGETGGSGGSGYTGHTGGSGGSGGSG
GSGTTGDVGLTGGTGSSTGETGGSGGSGGSGGSGYTGGSGYTGGTGGAILYTTVTVTSNYDVSGTNPVVKADGTLTVNLP
LANTKAIVRVINIGTGTVTVDPAGSQEVNGNVVHALTTQWQSATYVSDGSNWLV
If I move the FASTA file with this gene into my work directory,
mv /var/folders/gw/5mdblzs94gsb1ss44llgl3_h0000gn/T/tmpkldgr9mi/AA_gene_sequences.fa.0 gene.fa
and run this command (which is exactly what anvi'o runs here), I can confirm that it produces a segfault error:
hmmsearch -o gene_hmm_output.txt \
--cpu 1 \
--tblout gene_table.txt \
${anvio_path}/anvio/data/misc/KEGG/HMMs/Kofam.hmm gene.fa
Segmentation fault: 11
So there are this many lines in gene.fa:
wc -l gene.fa
49 gene.fa
If I cut it half into a shorter_gene.fa,
head -n 24 gene.fa > shorter_gene.fa
And then re-run the same command:
hmmsearch -o gene_hmm_output.txt \
--cpu 1 \
--tblout gene_table.txt \
${anvio_path}/anvio/data/misc/KEGG/HMMs/Kofam.hmm gene.fa
This time there is no segfault error, and I get my sweet sweet output files:
head gene_table.txt
# --- full sequence ---- --- best 1 domain ---- --- domain number estimation ----
# target name accession query name accession E-value score bias E-value score bias exp reg clu ov env dom rep inc description of target
#------------------- ---------- -------------------- ---------- --------- ------ ----- --------- ------ ----- --- --- --- --- --- --- --- --- ---------------------
29 - K06236 - 3.1e-17 47.6 228.3 7.3e-13 33.1 56.8 9.1 1 1 3 4 4 4 3 -
29 - K19470 - 1 -115.0 305.4 1.1e-06 14.2 8.7 16.9 15 3 2 18 18 18 0 -
29 - K03986 - 1.6e-09 22.8 68.6 1.8e-08 19.3 9.8 16.1 15 2 1 17 17 17 10 -
29 - K03987 - 6.5e-12 31.1 70.9 2.3e-09 22.7 6.7 15.6 15 1 1 16 16 16 5 -
29 - K22020 - 1.7e-12 30.2 187.1 7.1e-12 28.2 187.2 1.6 1 1 0 1 1 1 1 -
29 - K06237 - 1.2e-37 114.5 171.8 1.8e-17 47.6 55.3 5.3 2 2 1 3 3 3 3 -
29 - K03991 - 9e-10 24.1 19.1 9e-10 24.1 19.1 15.8 14 2 2 17 17 17 6 -
Contigs db with a single gene is here.
My HMMER version was this:
hmmscan -h 2>&1 | grep HMMER
# HMMER 3.3.1 (Jul 2020); http://hmmer.org/
I downgraded it to 3.2.1:
conda install -c bioconda hmmer=3.2.1
So the same output looked like this:
hmmscan -h 2>&1 | grep HMMER
# HMMER 3.2.1 (June 2018); http://hmmer.org/
Then running the same command worked:
anvi-run-kegg-kofams -c contigs.db --just-do-it
Modules database .............................: An existing database, /Users/meren/github/anvio/anvio/data/misc/KEGG/MODULES.db, has been loaded.
Kegg Modules .................................: 443 found
CITATION
===============================================
Anvi'o will annotate your database with the KEGG KOfam database, as described in
Aramaki et al (doi:10.1093/bioinformatics/btz859) When you publish your
findings, please do not forget to properly credit this work.
Contigs DB ...................................: Initialized: contigs.db (v. 20)
(...)
✓ anvi-run-kegg-kofams took 0:00:24.653547
thanks @meren and @ivagljiva!
did you still want the contigs db for the anvi-run-hmm issue to see whether there is an upstream problem in anvio or in the data? I have since figured out which one that is