Miss some metrics in airr.tsv file.
Here is the command: perl trust-airr.pl test1_barcode_report.tsv test1_annotate.fa --format barcoderep > airr_rearrangement.tsv
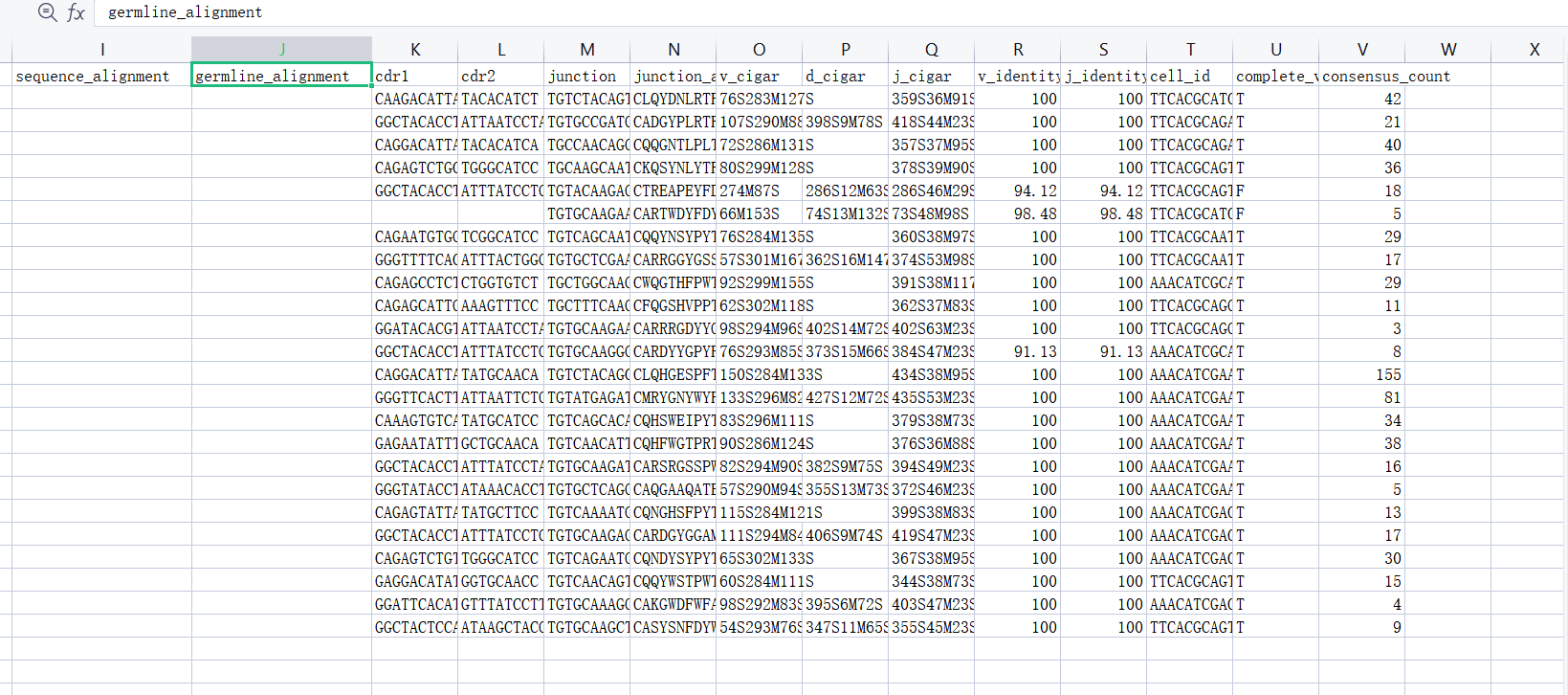
As the figure shown, sequence_alignment, germline_alignment, cdr3nt, cdr3aa, and location of cdr1 cdr2 cdr3 are not in airr file.
sequence_alignment, germline_alignment output is implemented in the github repo. I will draft a new release this week. "cdr3nt, cdr3aa, location of cdr1,2,3" are optional fields in the airr file, so I did not output them. In AIRR specification, "junction" is the cdr3 with flanking motif amino acids, so they are kind of equivalent. Hope this helps.
If you want to generate the "sequence_alignment, germline_alignment" fields directly from trust-airr script, you need to return annotator with option "--airrAlign" to output the two alignment information, and feed that file to the "--airr-align" option of the trust-airr.pl script.

Yes. I did not see --airrAlignment option, so you need to use "git clone" to get the github version.
Ok, i will try.
Another question is how to calculate the umi for each contig? I found the umi count is equal to read count after converting barcode report to 10x.

The UMI information is just used for abundance estimation, that's why you see the read count (abundance) equals to umi when using the "--UMI" option.
So, When using --UMI option, read count is an accurate read count value, and represents the abundance, but the umi is just a abundance metric, not a real umi count value in the 10X report?
Sorry for my bad explanation. When using --UMI option, read count will be computed as UMI count instead of the actual read count.
Thanks. so how to calculate the real read count for each assembled contig?
If you want to the abundance using read count, you can run TRUST4 without the UMI information.