zgtools-pipeline
 zgtools-pipeline copied to clipboard
zgtools-pipeline copied to clipboard
zgtools-pipeline: A pipeline that allows for the convenient acquisition of T2T (Telomere-to-Telomere) genomes.

zgtools-pipeline
-
zgtools是一款自主编写的模块集合软件,其中包含了超过100个模块用于处理常规基因组与T2T基因组,其涵盖Survey、组装、HIC、T2T、注释、进化分析、着丝粒预测以及多种个性化分析功能。该软件旨在通过一个统一的启动器来管理所有基因组分析任务,相较于nextflow流程,它提供了更高的灵活性。
-
zgtools结合一百多个基因组的运行经验,对许多标准分析流程进行了优化,并实现了自动化更新,其目标是实现“更快、更准、更好”的分析效果。
-
目前zgtools已经完成了十多个物种的T2T组装,并将相关成果发表在了HR、NC等期刊上。对于绝大部分植物、哺乳动物以及鱼类(包括含有ZW/XY性染色体的物种),zgtools能够轻松实现T2T组装,并提供完整的PAR(PAR指“伪常染色体区域”)。
-
对于300M至1G大小的基因组,从基因组的Survey、组装到HIC的整个流程,zgtools只需3至7天即可完成。同样地,对于300M至1G大小的基因组,从T2T组装、Gap填补、端粒延伸修补到评估的整个流程,zgtools也只需7至14天。
-
此外,这是一个【个人项目】,目前暂不开源,但也提供科研咨询服务。其他方面,如果您需要HIC调图教程或测序服务,也可通过QQ【1954616586】与我联系。
目录
- zgtools-pipeline
- 流程图
- 测序咨询
- 完成度
- 分析内容
- 最近更新
- 运行教程
流程图
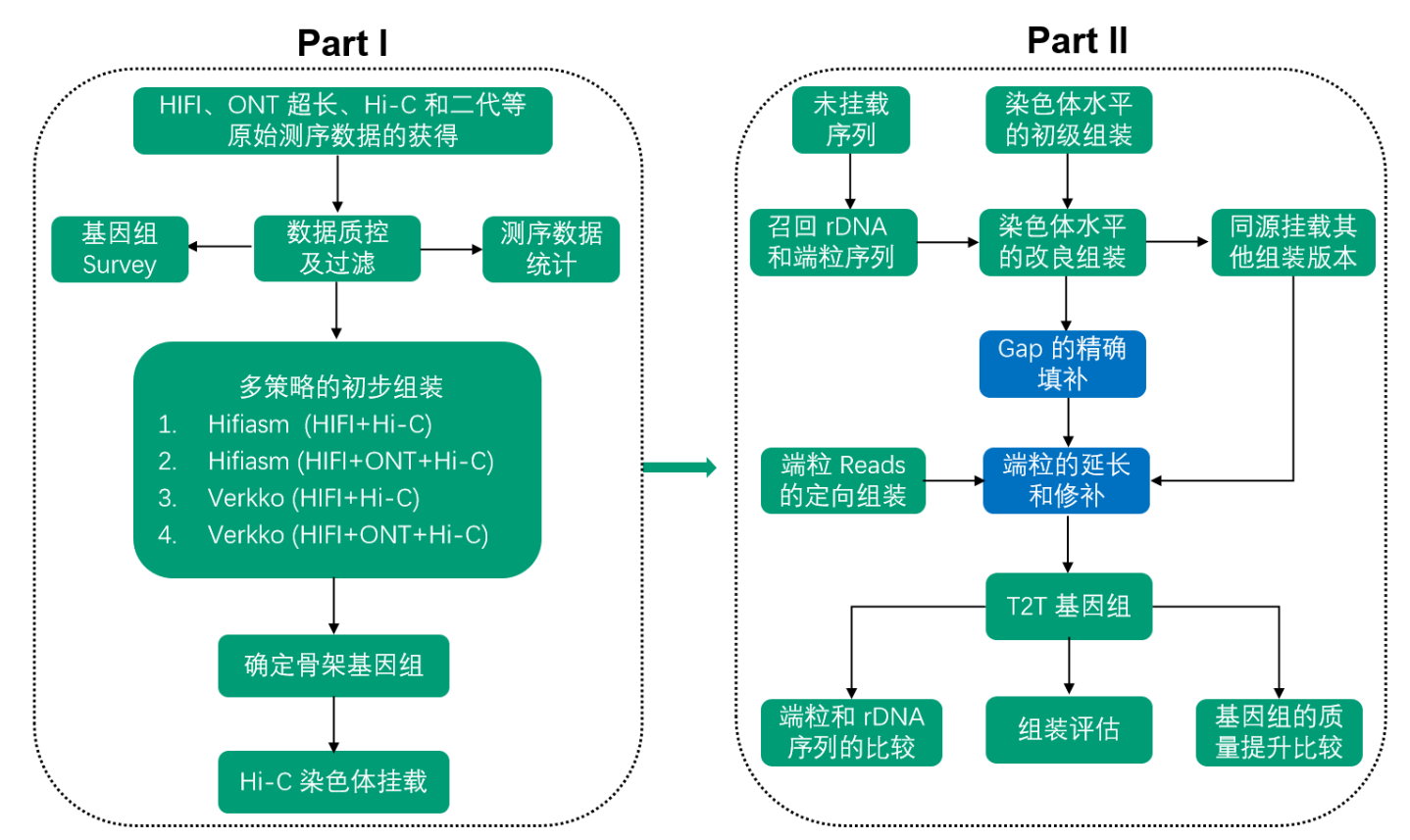
T2T组装流程图:
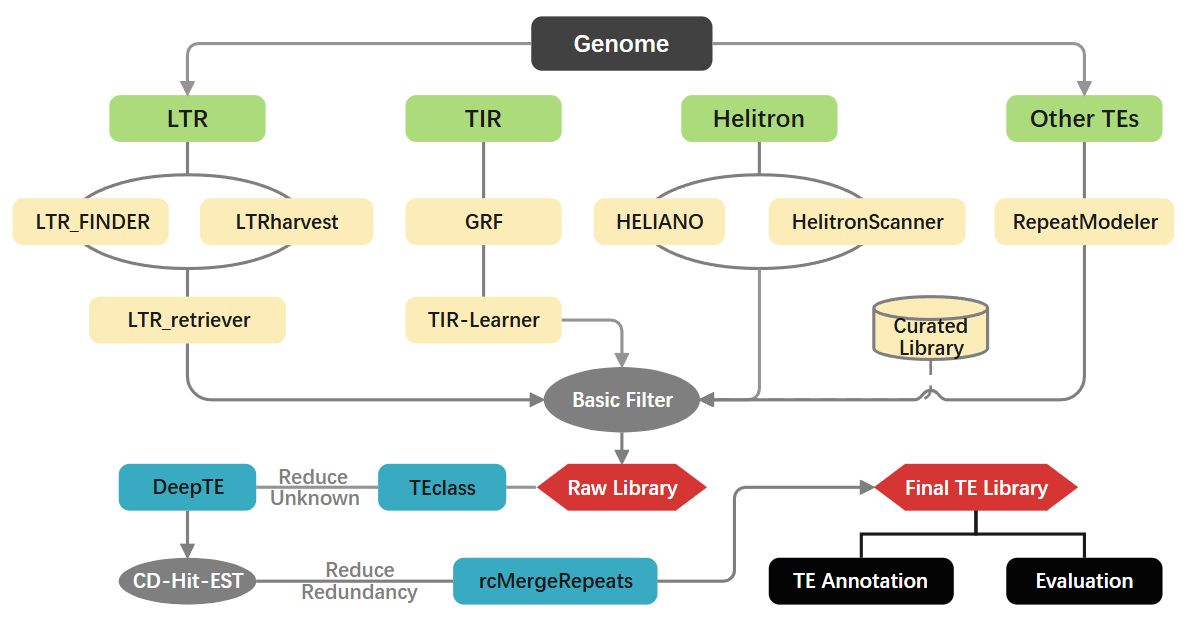
重复序列注释流程图:
测序咨询
①HIFI数据,12000一个Cell,保底80g,一般可以测到90g;如果需要测少量的HIFI,即散测。散测费用是建库2000,测序180/g;
②HIC数据,一个文库4000,测序10/g;
③ONT数据,默认N50:100K,目前11000一个Cell,单个Cell产出植物15G,哺乳动物20G以上,水产和昆虫这些暂不承诺;
④二代数据:提取建库110,测序10/G;

完成度
zgtools达到的T2T水平:
①0 Gap:最基本的要求;
②全端粒:每条染色体末端端粒重复次数大于100次(一般1000次以上比较好);
③全rDNA:有rDNA末端的染色体也完善出端粒;
④全着丝粒:整个基因组准确鉴定着丝粒。
结果文件请见示例【example】,要求作图与结果均达到CNS水平。
分析内容
01、T2T端粒延伸+Gap填补+T2T纠错+T2T各项评估
02、Survey分析+倍型分析+二代/三代NT比对去污染
03、常规基因组组装/分型基因组组装/T2T基因组组装(端粒延伸、补Gap、全rDNA和动物T2T-Y染色体的PAR区)+各项评估
04、HiCUP评估(HiC小测/HiC大测)+HiC挂载调图+染色体级别基因组生成+HiC热图+未挂载区去冗余+共线性分析+各项评估
05、Subphaser亚基因组分型+亚基因组特有分析(SV分析/等位基因表达不平衡分析/亚基因组优势分析/KaKs差异分析)
06、LAI评估+LTR插入时间分析+LTR-RTs系统发育树(最大似然法)
07、着丝粒预测(优于目前已发表的所有生信鉴定着丝粒软件)
08、重复基因鉴定+KaKs分析+功能富集
09、圈图、基因共线性
10、注释与进化(待更新)
11、多倍体T2T流程(更新中)
最近更新
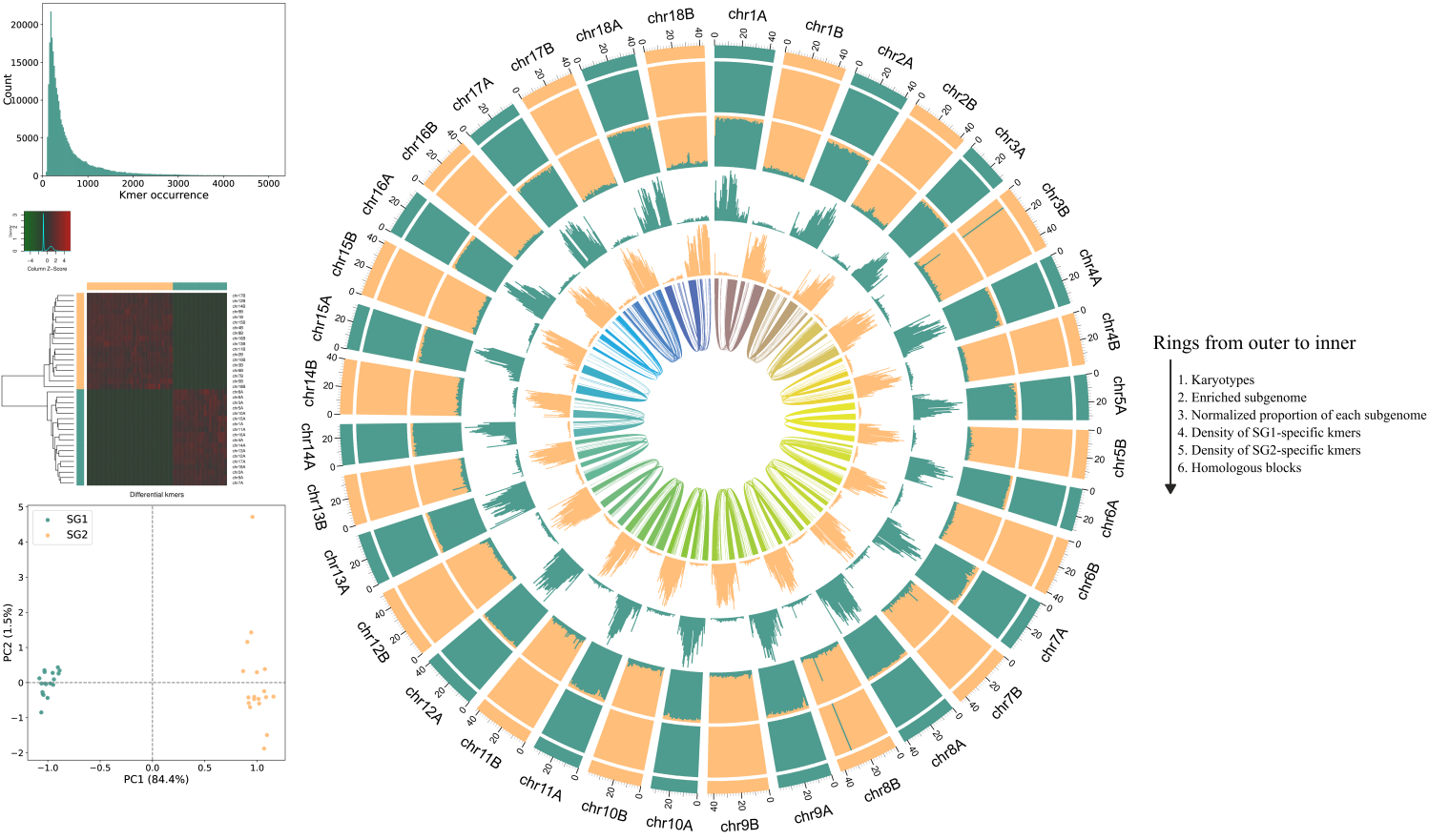
☆2025/1/7---亚基因组分型:subphaser(亚基因组分型)
运行zgtools subphaser进行分析,填写配置文件运行即可,原软件SubPhaser写的很好,我们只在原基础上修改了配色和添加图例,结果如下:
☆2024/12/24---候选着丝粒可视化:plotCCR(展示候选着丝粒区域)
运行zgtools plotCCR进行分析,检查重复序列的分布得到候选着丝粒区域,结果如下:

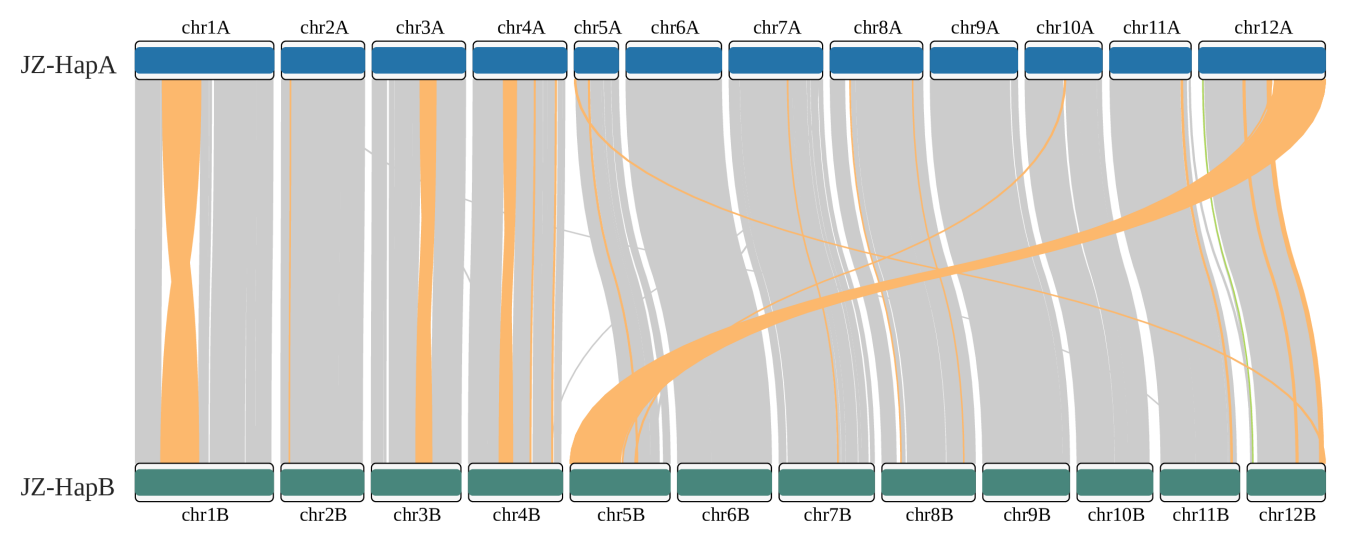
☆2024/12/05---染色体SV可视化:GetTwoSyn(展示染色体间的SV)
运行zgtools GetTwoSyn进行syri的切分并行分析,展示基因组共线性(基因共线性建议JCVI)和染色体间的SV:INV(黄色)、TRANS(绿色)和 Dup(蓝色)。以【T2T骏枣 】的chr5和chr12染色体的移位为例,结果如下:
基因共线性圈图:
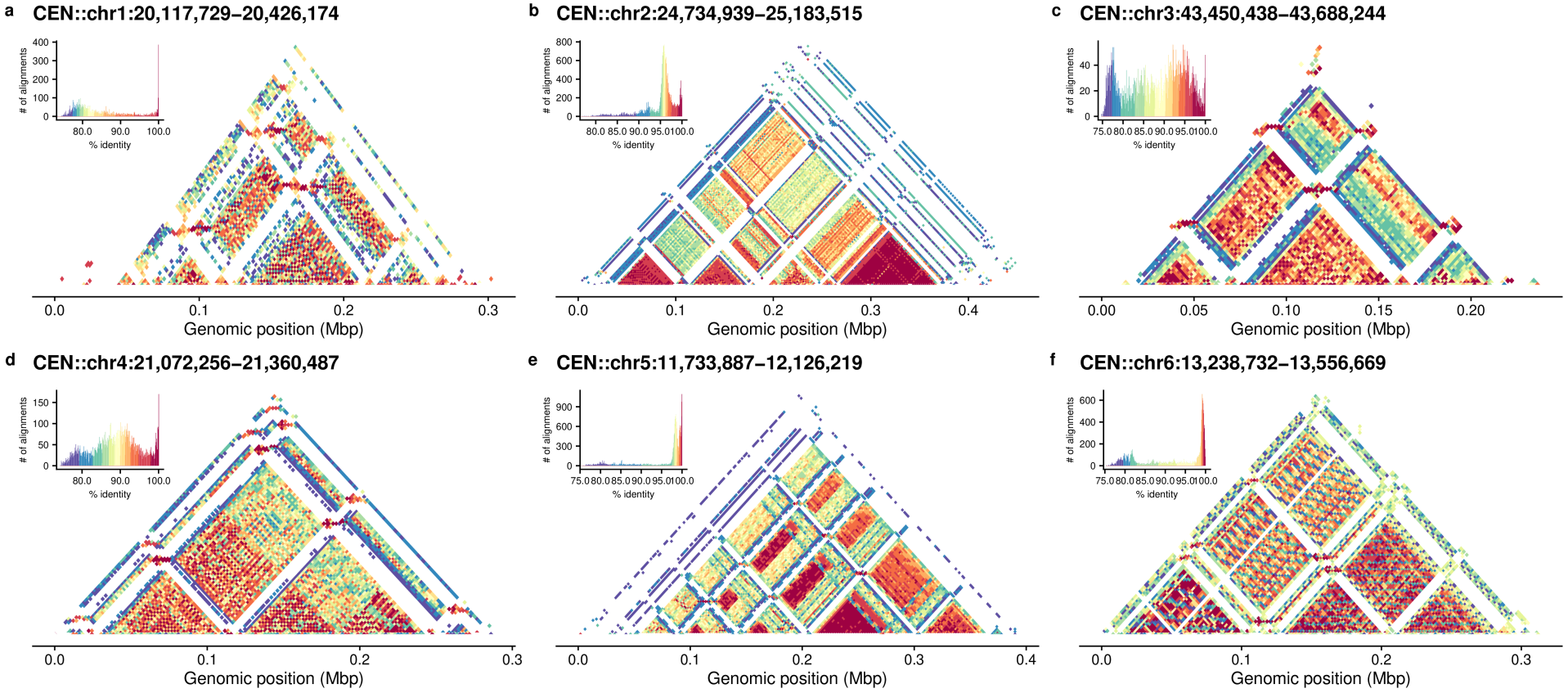
☆2024/12/03---着丝粒可视化:StaniedGlass(提速与并行)
运行zgtools run_centromere获得着丝粒的候选区域后,可运行zgtools StainedGlass进行可视化,该模块在原代码的基础上进行修改,加速运行而支持更方便的并行,对于结果文件也进行了精简,对于着丝粒区域的可视化运行速度大大提升。示例结果如下:
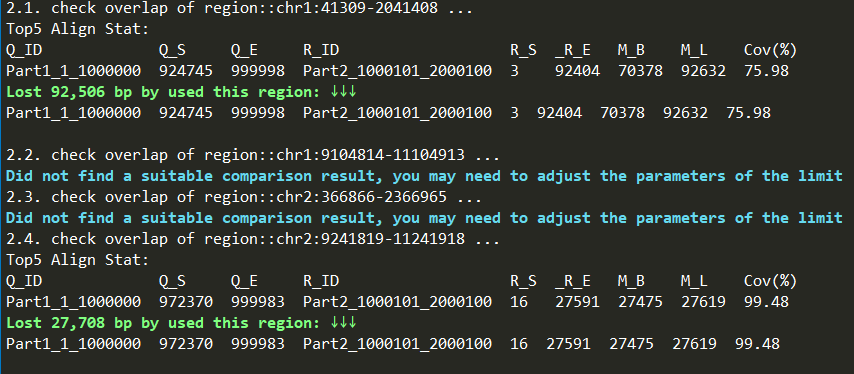
☆2024/12/03---Gap填补:gapjoin(Gap拓展区域重复检测)
使用zgtools mdifgap进行多重数据填补Gap后,仍有填补不上的结果,通过Juicebox检查发现,Gap的邻近区域存在600k的冗余序列,故而仅依靠Reads是无法填补的,由此引入zgtools gapjoin模块,通过对Gap区域左右拓展一定区域,对该区域进行比对检查是否存在重复的部分。筛选高Coverage和长比对区域并限制基因组损失的极限值,将Gap区域附近的冗余删除进而连接起来,实现填补Gap的目的,最后绘制Reads覆盖图进一步检查。示例如下:
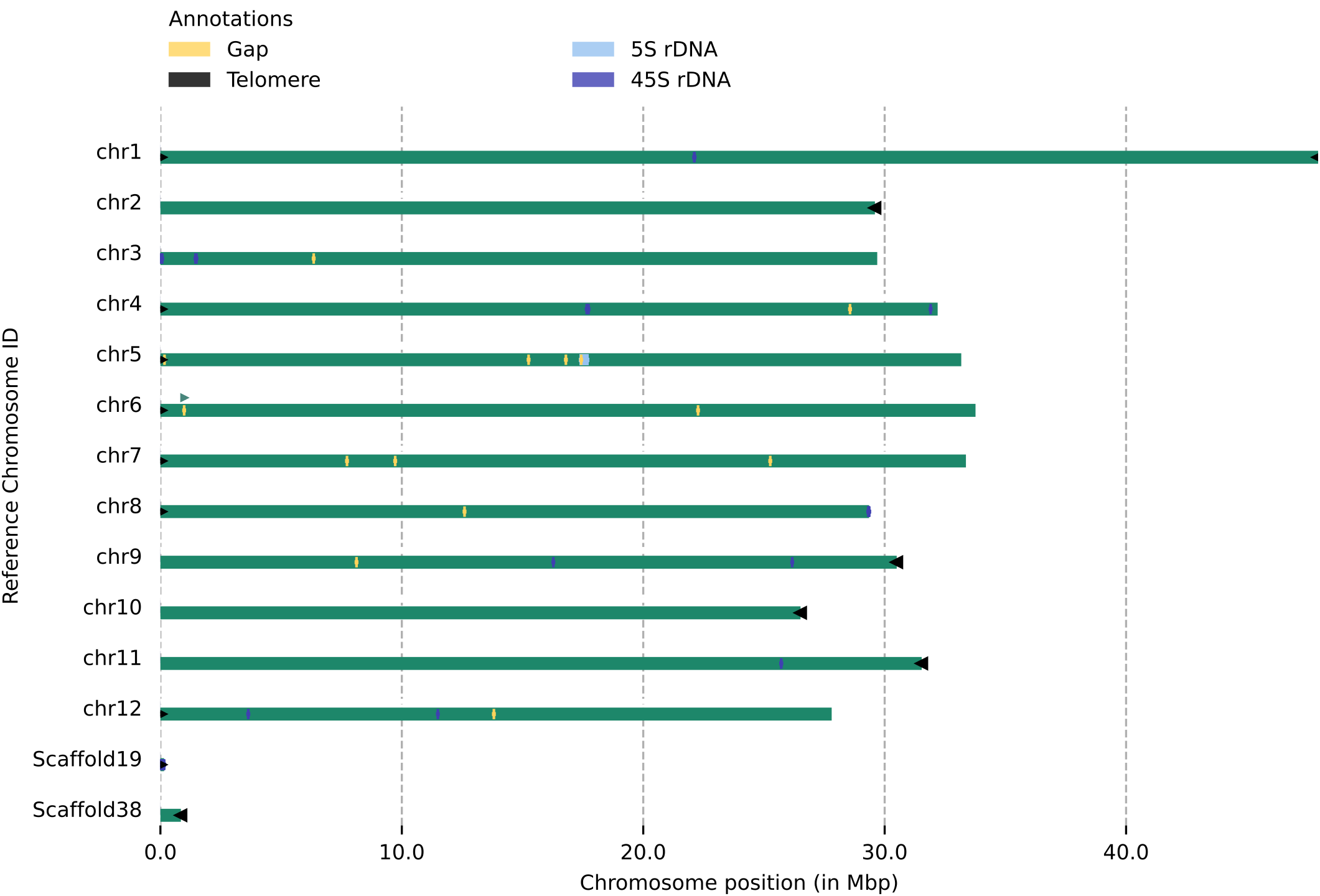
☆2024/12/02---端粒、rDNA、Gap检查:genomecheck(端粒遮掩检测)
使用zgtools genomecheck对基因组(包含未挂载区的序列)进行端粒、Gap和rDNA的鉴定,由此可以①获得基因组的染色体部分的初步端粒情况;②结合未挂载区的端粒与rDNA,可以回捞端粒回染色体部分,比如示例图中的Scaffold19可以通过检查Juicebox图加回至chr3上游的45rDNA末端或者chr8下游的45rDNA末端;③检测是否有端粒序列被其他序列遮挡住,如图中chr6上游的遮掩标识(绿色三角形)表示附近的序列被邻近的序列遮挡,返回Juicebox检查发现前一序列是该序列的冗余部分。
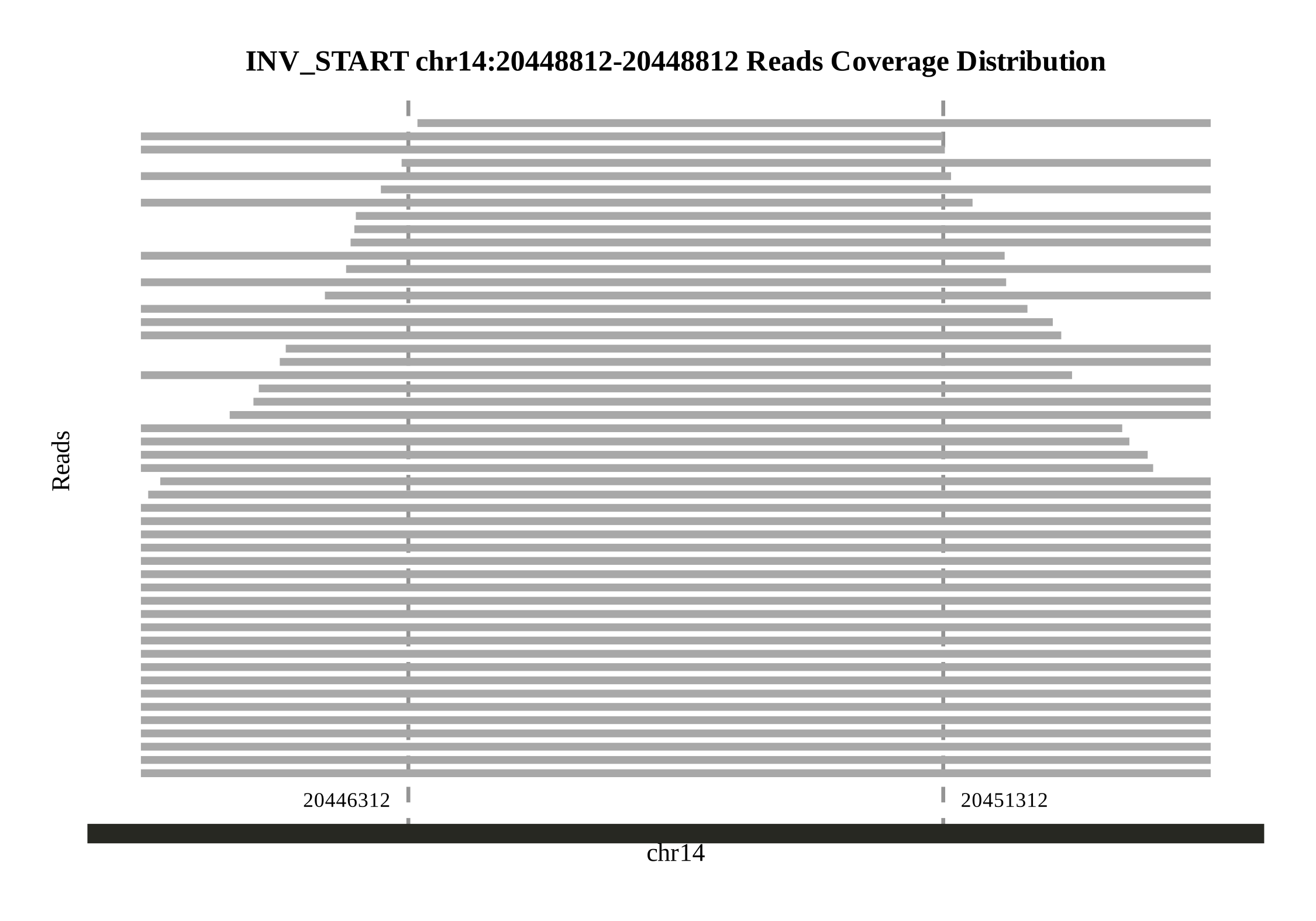
☆2024/11/28---SV断点自动检查:run_CheckSV(Reads覆盖图)
首先,使用zgtools工具集中的syntenic和genomesyn功能进行共线性分析。这一过程将产生基因组共线性图以及名为Ref_vs_Query.syri.out的变异检测结果文件。随后,对SV(结构变异)进行长度过滤,仅保留长度大于500,000bp的SV,并从中提取SV的起始和终止位置信息。接着,利用minimap2工具将HIFI数据和ONT数据回比到基因组上。在此基础上,我们对已确定的SV位置进行左右两侧各拓展5,000bp的处理。之后,批量绘制这些区域的Reads覆盖图,并自动检测Reads的覆盖情况。示例图如下:
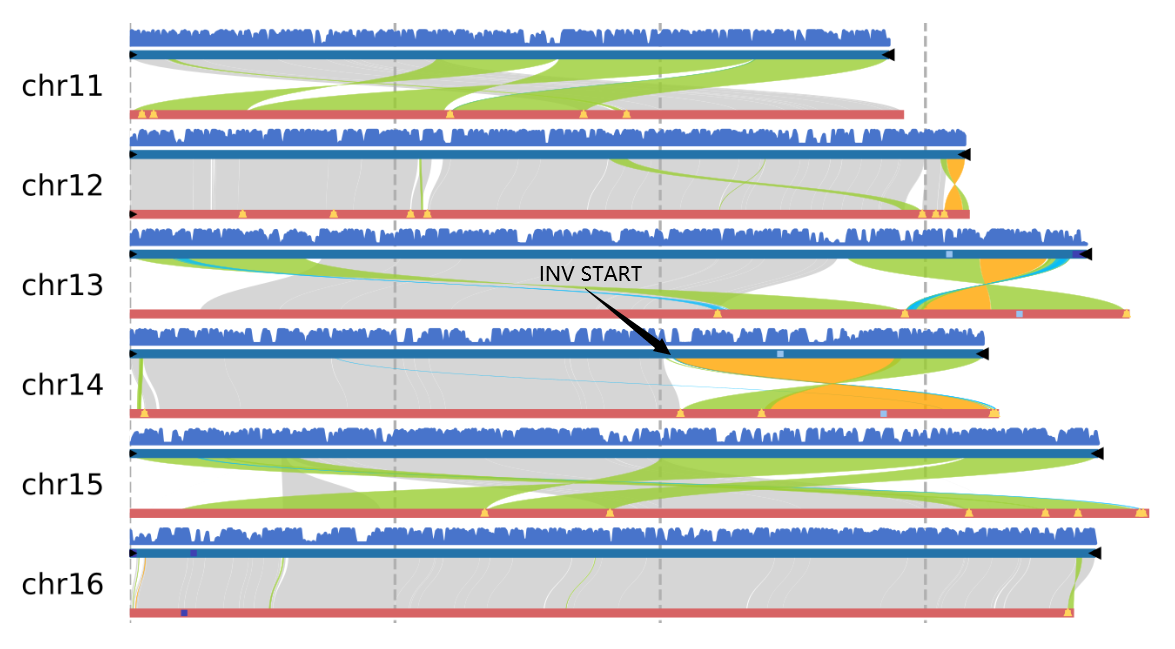
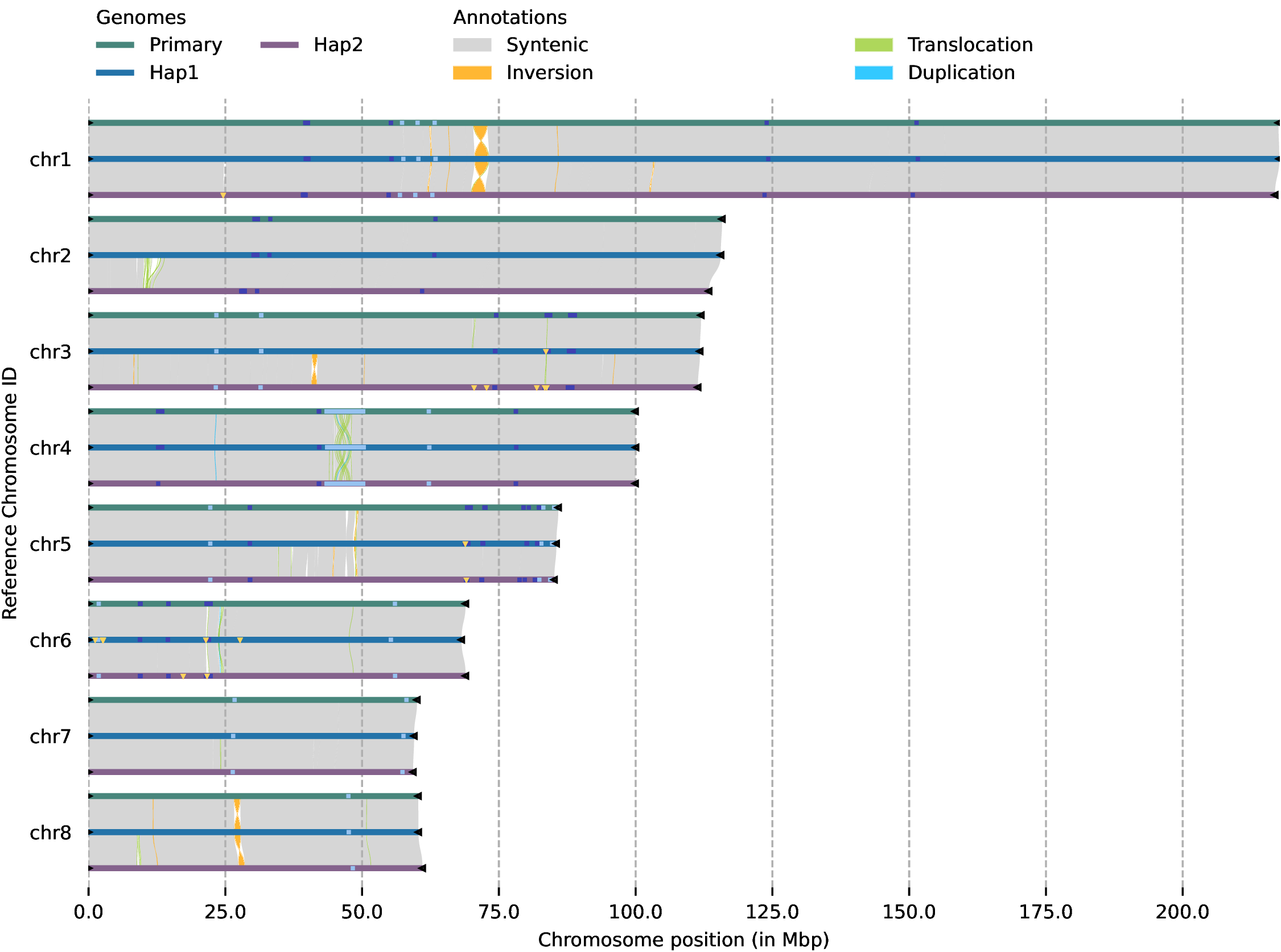
☆2024/11/22---共线性图:ngenomesyn(多基因组)
多基因组共线性图,无数量上限,图中黑三角为端粒,黄三角为组装的 Gap 序列(T2T基因组则没有),浅蓝色为 5S_rDNA,深蓝色为 45S_rDNA,Syntenic 为共线性区域,Inversion 为倒位区域,Translocation 为易位区域,Duplication为重复区域。
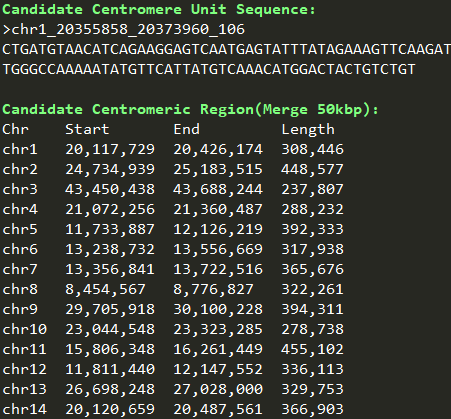
☆2024/11/22---着丝粒预测:run_centromere(更新)
测试:对TR富集或者LTR富集的植物,以及TR和LTR都比较少的鱼类进行测试,结果很不错,相较于quarTeT和CentIER都有很大的提升:使用TRF软件鉴定串联重复序列,筛选重复次数>100,单元长度>100bp的TR序列,基于社区检测算法,以80%相似度进行构建TR相似性网络,对筛选的单体序列进行重鉴定,对TE和TR高覆盖的区域进行筛选整合,得到着丝粒核心区和近着丝粒区,以下是其中鱼类的测试结果:
☆2024/11/18---Gap填补:mdifgap(多重数据迭代填补)
测试:500M基因组17个gap和2G基因组5个gap,分别用时13分钟和5分钟,填补速度非常快,消耗内存非常小,有补gap绘制reads覆盖图检查HIC调图是否有问题和补gap后对新区域进行reads验证检查是否有问题。目前来说其他软件:
①TGS-Gapcloser很容易给基因组补出许多序列,补gap前后可能多出好几M,其次缺点还有就是容易爆内存。
②quarTeT的补gap也有问题,效果差,补完gap,多出100多M也不说了。如果实在要补gap还是用TGS-Gapcloser吧,慢慢跑,慢慢补应该不会爆内存。其他的例如,LR-gapcloser也效果不是很好。
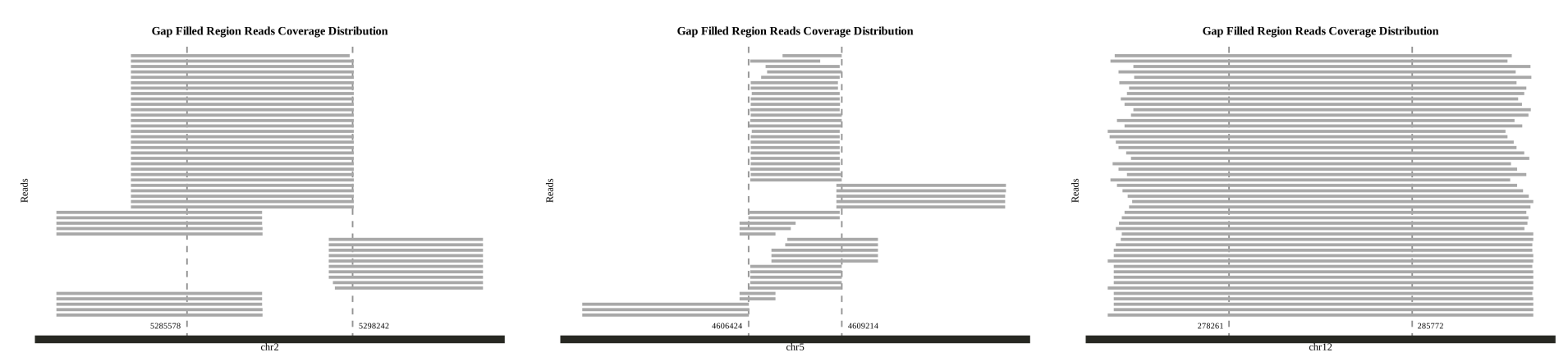
使用 winnowmap2/minimap2 将补洞数据(不含N)与基因组 Gap 区间比对(含N),该步骤分为三个水平对 Gap 进行填补,其优先级为:其他基因组版本>HIFI数据>ONT数据。①其他版本基因组为组装的各版本ctg级别基因组,除primary嵌合体以外,还包括hap1和hap2的基因组;②HIFI数据即环化后的 HIFI 数据;③ONT数据可以为经过纠错后的一致性序列,比如nextdenovo生成的cns序列,也可以是经过HERRO模型纠错后的R10数据。zgtools mdifgap将会进行每种数据做三轮Gap填补,最多进行9轮Gap填补。
左图为ONT数据填补的结果;中间图为HIFI数据填补的结果;右图为其他组装版本填补的结果。如图所示,由于HIFI数据一般比较短,在一些Gap区域,往往需要通过“搭桥”的形式通过Gap替换区域的两端,对于一些复杂的Gap区域,ONT序列也可能需要“搭桥”通过。最理想的情况为右图,原Gap周围没有复杂的冗余与未切断序列,可以被很好的填补上。
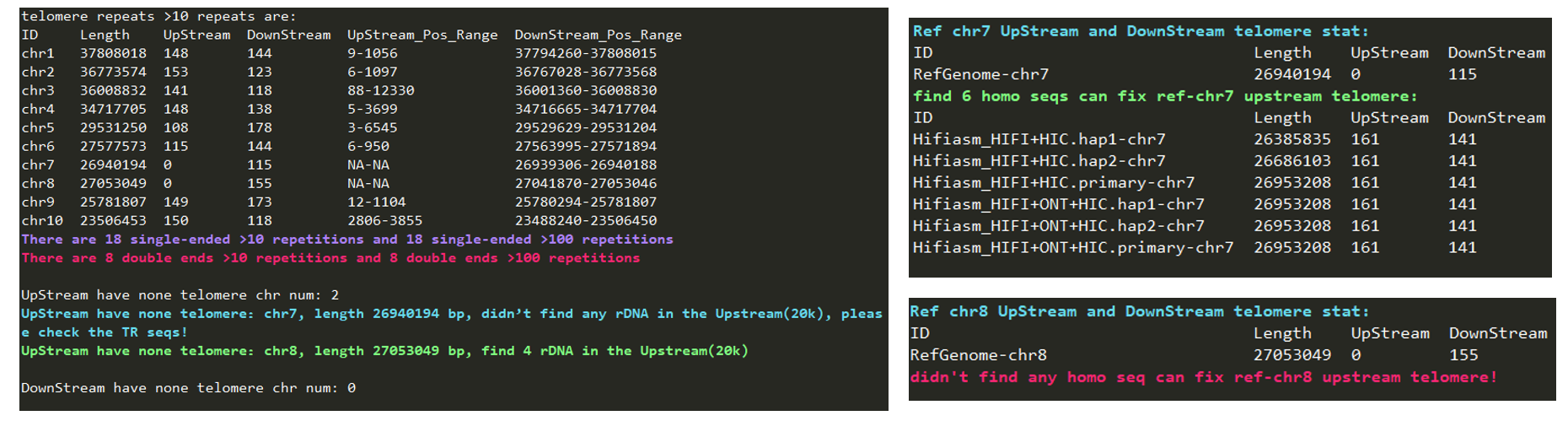
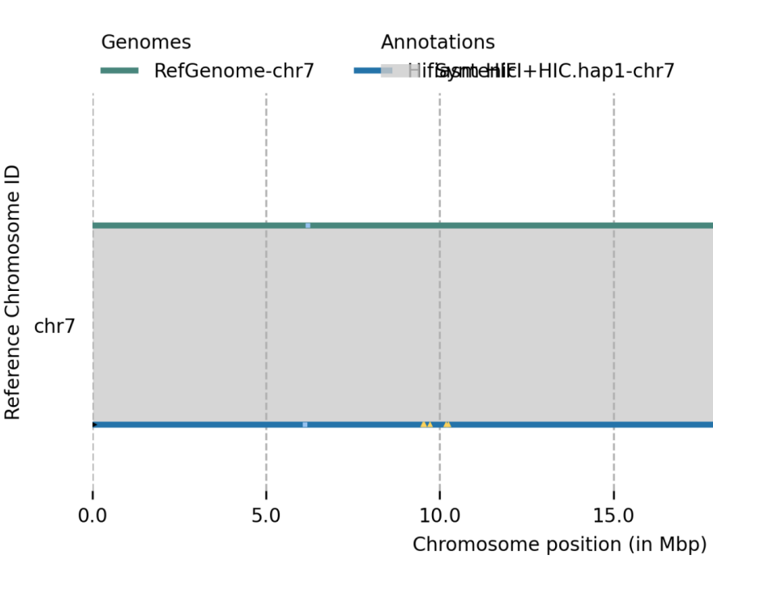
☆2024/11/18---端粒修补:homotelo(同源比较)
获得 0 Gap的基因组后通过端粒鉴定,发现端粒有不完整的时,开始进行端粒修补,此时,使用homotelo模块,原理是将其他组装版本与0 Gap基因组进行比较,基于高质量的共线性与其他限制性参数对基因组末端进行末端替换,homotelo的末端替换不只限于端粒的替换,还能够检查当前T2T骨架版本是否有未装出来的部分,生成.hic文件,导入Juicebox验证准确性。如果还有修补失败的末端,再使用zgtools homorDNA和zgtools telofix进行rDNA与三代数据的端粒延伸。
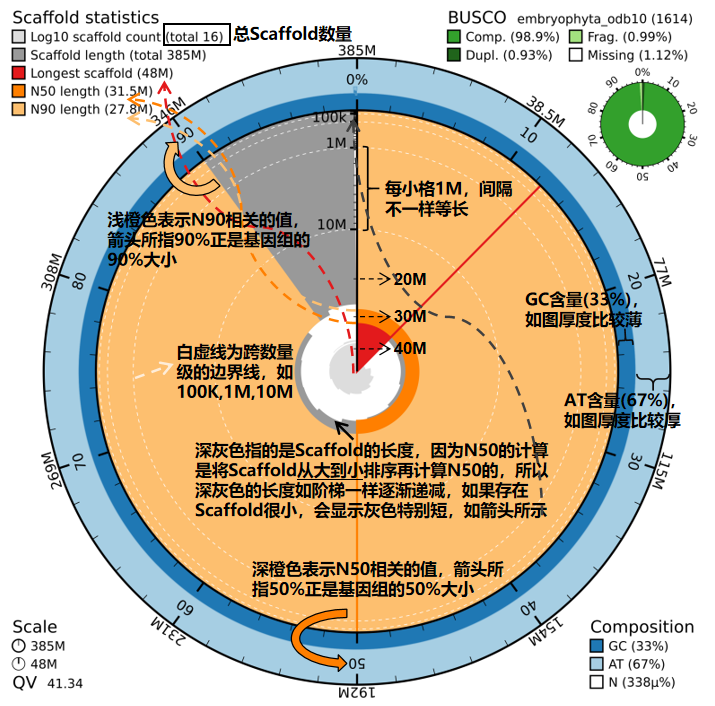
☆2024/11/18---蜗牛图:snailplot
蜗牛图(Snail Plot),其主要是根据 Scaffold 的(或 Contigs)N50/N90 指标,QV 评估结果以及 BUSCO 评估结果,对基因组组装质量进行直观的可视化展示,结果如下图所示:
绘制端粒/rDNA/Gap预览图如下:

注: 端粒/rDNA分布预览图中, 末端的绿色为端粒基序重复次数大于100的端粒, 深蓝色为45 rDNA, 浅蓝色为5S rDNA, 红色为Gap.
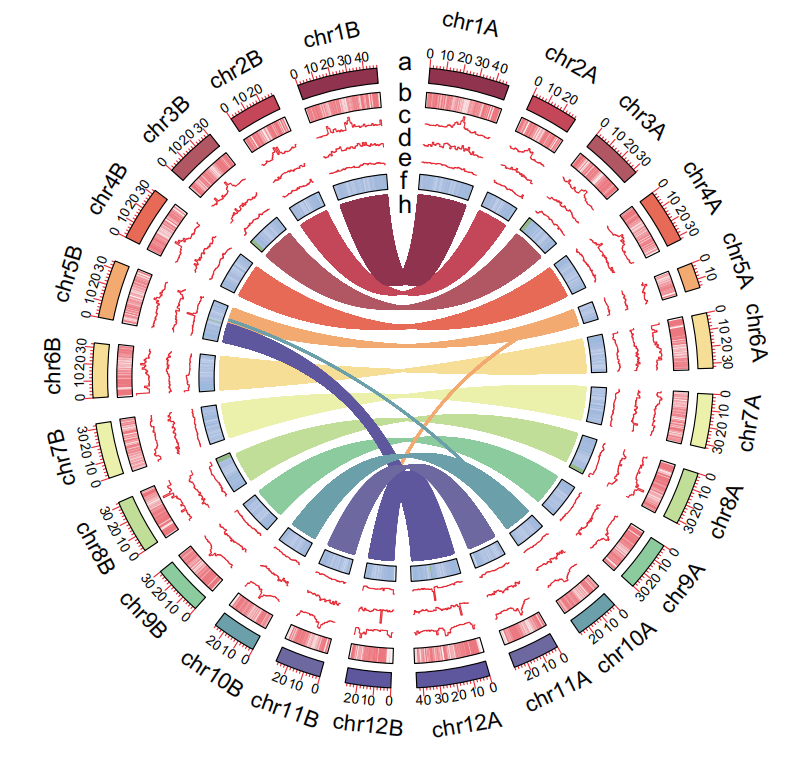
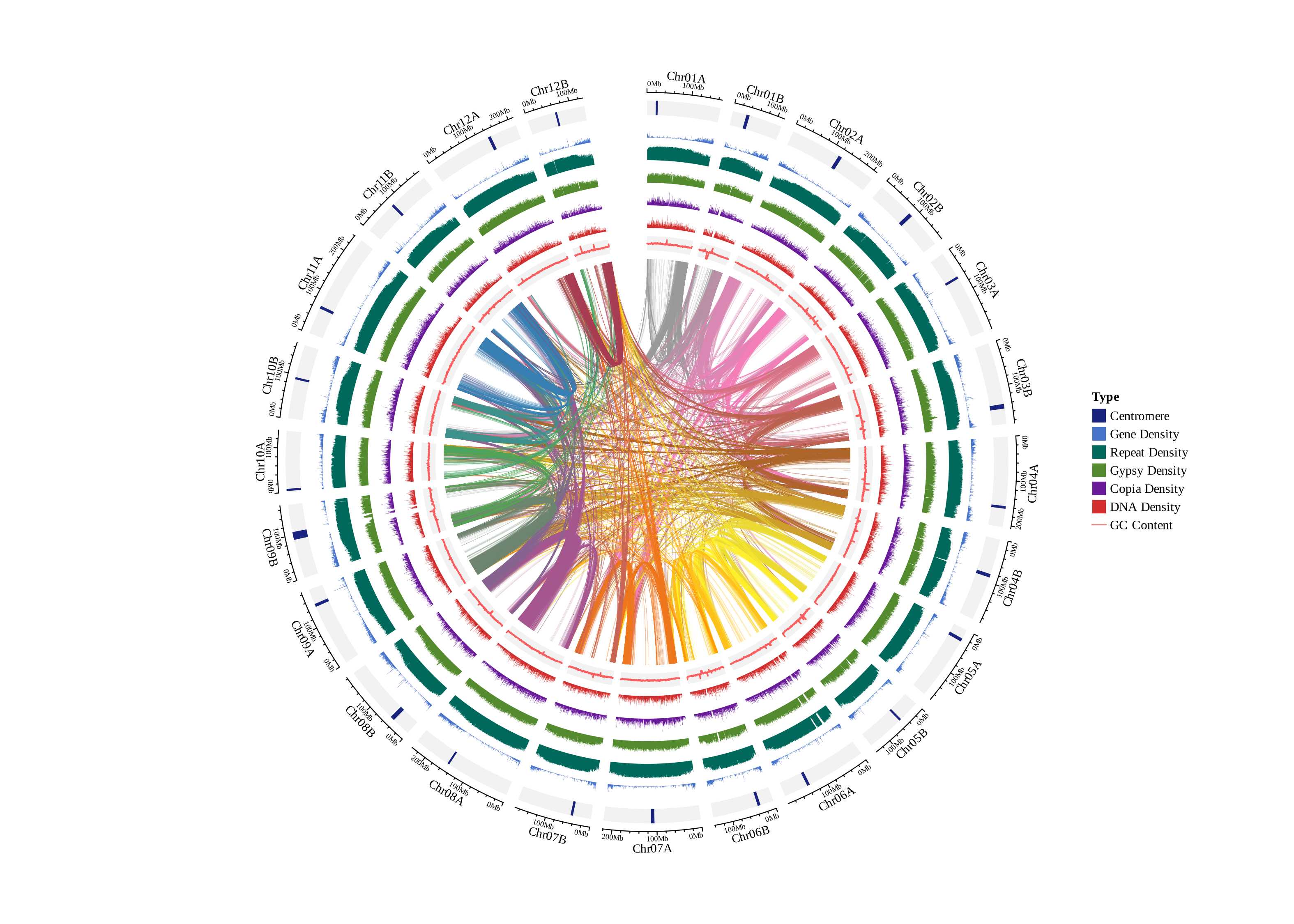
☆T2T圈图
①展示所有共线性
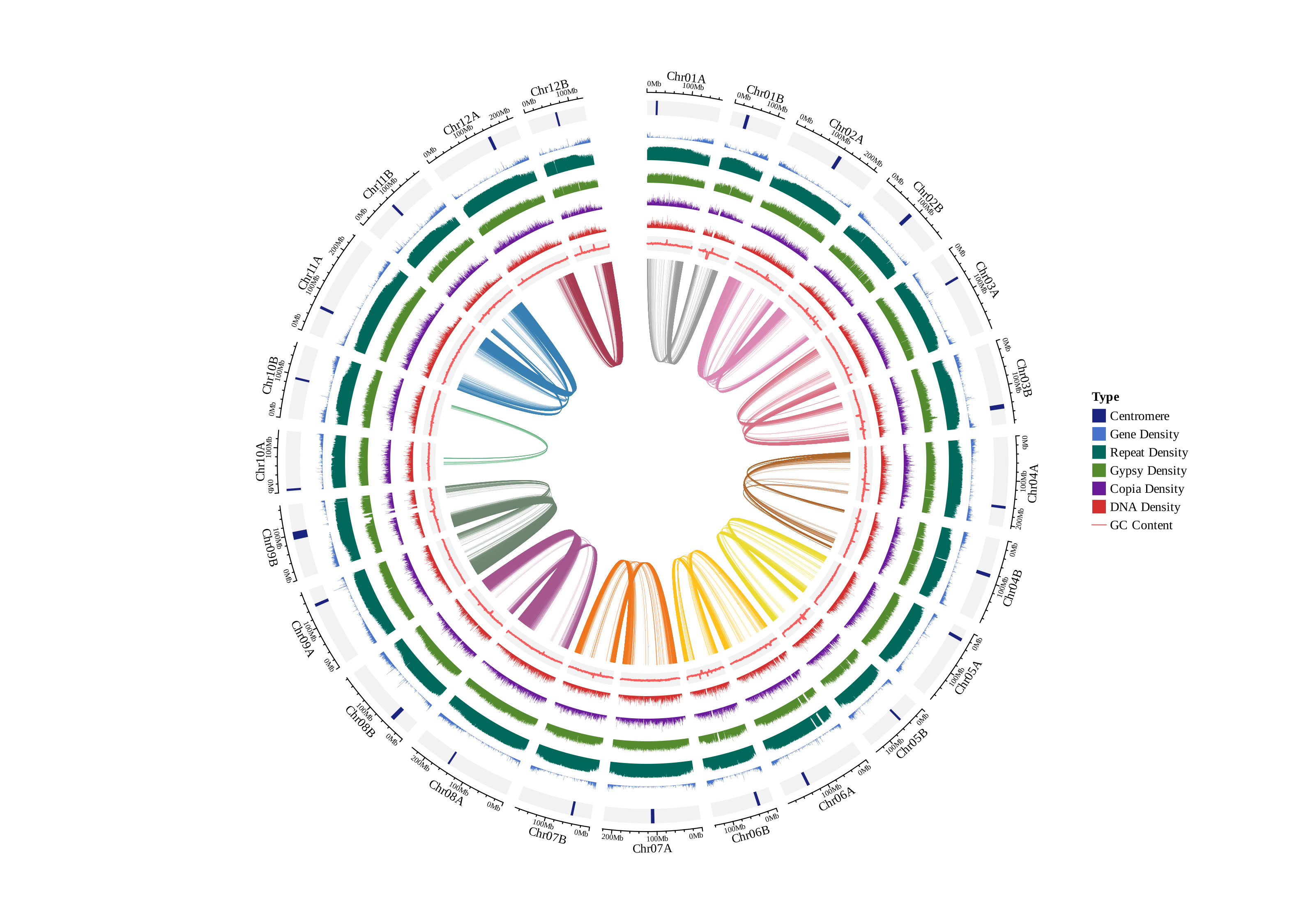
 ②ID保持邻近,只展示Chr01A_vs_Chr01B,...的共线性
②ID保持邻近,只展示Chr01A_vs_Chr01B,...的共线性
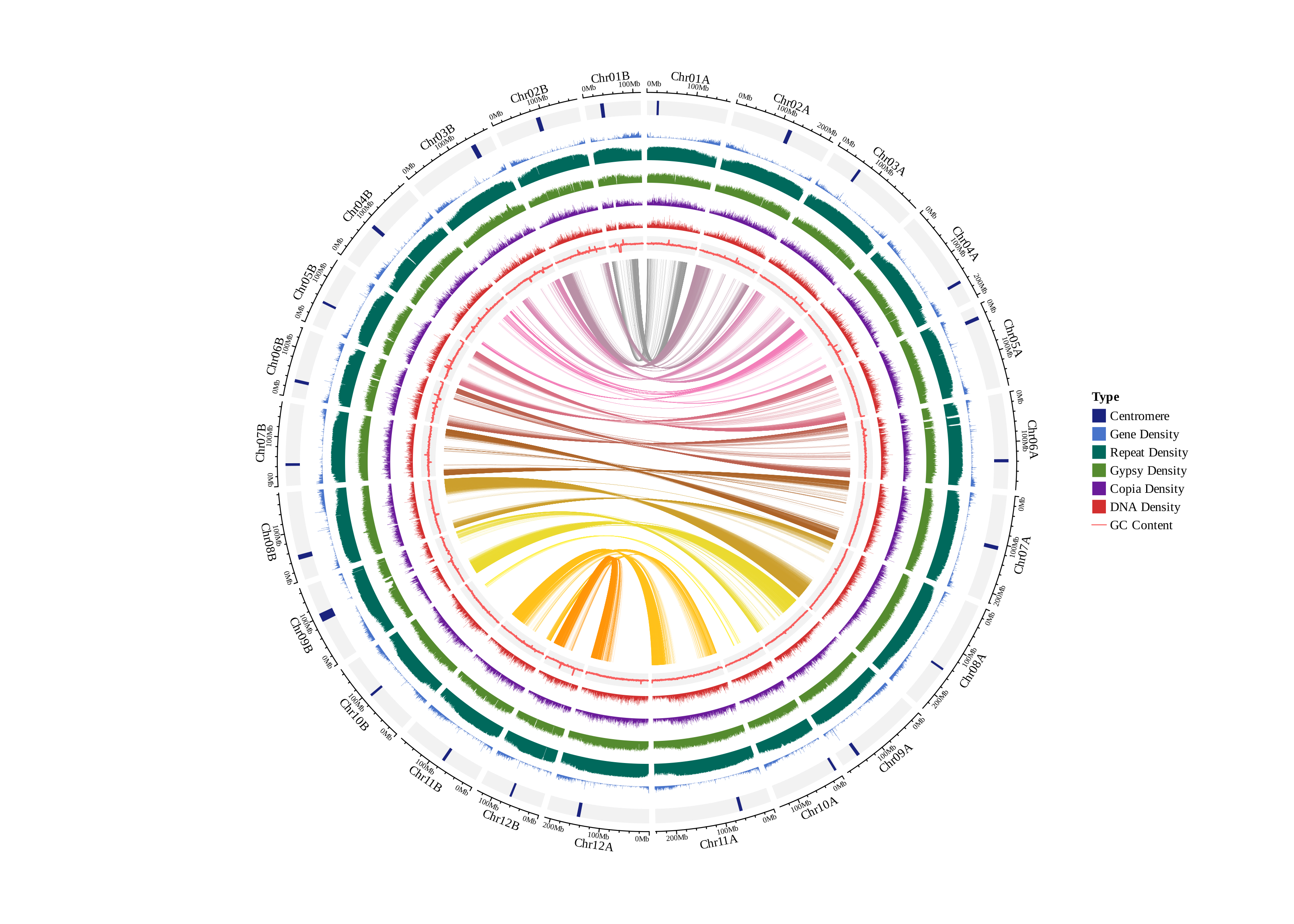
 ③Y轴对称(当基因组为异源四倍体时,则不会Y轴完全对称),只展示Chr01A_vs_Chr01B,...的共线性
③Y轴对称(当基因组为异源四倍体时,则不会Y轴完全对称),只展示Chr01A_vs_Chr01B,...的共线性
运行教程
后续更新...