ComplexHeatmap
 ComplexHeatmap copied to clipboard
ComplexHeatmap copied to clipboard
Error in quantile.default(abs(mat), 0.975) : missing values and NaN's not allowed if 'na.rm' is false?
I am not sure why I am getting this error
Could you please help me with any info so that I could move forward? 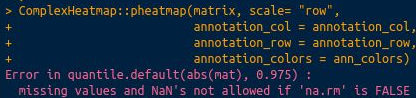
I attached the ps.prev0 here: ps.prev0.zip
my codes
# Species ----
ps.prev0.Species.no.na <- ps.prev0 %>%
subset_taxa(!is.na(Species)
)
ps.prev.taxa <- tax_glom(ps.prev0.Species.no.na, taxrank = "Species", NArm = TRUE)
ps.taxa.pse <- ps.prev.taxa
otu_table(ps.taxa.pse) <- otu_table(ps.prev.taxa) + 1
sample_data(ps.taxa.pse) # sanity check
# pairwise comparison between 2wk and 14wk post 1st feeding
deseq = phyloseq_to_deseq2(ps.taxa.pse, ~ dev.stage)
# converting counts to integer mode
# Warning message:
# In DESeqDataSet(se, design = design, ignoreRank) :
# some variables in design formula are characters, converting to factors
ds = DESeq(deseq, test="Wald", fitType="local")
alpha = 0.05
res = results(ds, alpha=alpha)
res = res[order(res$padj, na.last=NA), ]
taxa_sig = rownames(res[1:20, ]) # select bottom 20 with lowest p.adj values
ps.taxa.rel <- transform_sample_counts(ps.taxa.pse, function(x) x/sum(x)*100)
ps.taxa.rel.sig <- prune_taxa(taxa_sig, ps.taxa.rel)
matrix <- as.matrix(data.frame(otu_table(ps.taxa.rel.sig)))
colnames(matrix) <- as.character(tax_table(ps.taxa.rel.sig)[, "Species"])
metadata_sub <- data.frame(sample_data(ps.taxa.rel.sig))
# Define the annotation color for columns and rows
annotation_col = data.frame(
dev.stage = as.factor(metadata_sub$dev.stage),
check.names = FALSE
)
rownames(annotation_col) = rownames(metadata_sub)
annotation_row = data.frame(
Phylum = as.factor(tax_table(ps.taxa.rel.sig)[, "Phylum"]),
check.names = FALSE
)
rownames(annotation_row) = colnames(matrix)
# ann_color should be named vectors
phylum_col = RColorBrewer::brewer.pal(length(levels(annotation_row$Phylum)), "Paired")
names(phylum_col) = levels(annotation_row$Phylum)
ann_colors = list(
dev.stage = c(`dev.stage-1` = "red", `dev.stage-2` = "blue"),
Phylum = phylum_col
)
ComplexHeatmap::pheatmap(matrix, scale= "row",
annotation_col = annotation_col,
annotation_row = annotation_row,
annotation_colors = ann_colors)
sessionInfo()
# R version 4.1.0 (2021-05-18)
# Platform: x86_64-conda-linux-gnu (64-bit)
# Running under: Ubuntu 20.04.4 LTS
#
# Matrix products: default
# BLAS/LAPACK: /home/r01mt19/.conda/envs/updatedR/lib/libopenblasp-r0.3.18.so
#
# locale:
# [1] LC_CTYPE=en_GB.UTF-8 LC_NUMERIC=C LC_TIME=en_GB.UTF-8 LC_COLLATE=en_GB.UTF-8
# [5] LC_MONETARY=en_GB.UTF-8 LC_MESSAGES=en_GB.UTF-8 LC_PAPER=en_GB.UTF-8 LC_NAME=C
# [9] LC_ADDRESS=C LC_TELEPHONE=C LC_MEASUREMENT=en_GB.UTF-8 LC_IDENTIFICATION=C
#
# attached base packages:
# [1] parallel stats4 stats graphics grDevices utils datasets methods base
#
# other attached packages:
# [1] DESeq2_1.32.0 SummarizedExperiment_1.22.0 Biobase_2.52.0 MatrixGenerics_1.4.0
# [5] matrixStats_0.61.0 GenomicRanges_1.44.0 GenomeInfoDb_1.28.0 IRanges_2.26.0
# [9] S4Vectors_0.30.0 BiocGenerics_0.38.0 vegan_2.5-7 lattice_0.20-45
# [13] permute_0.9-5 ggpubr_0.4.0 agricolae_1.3-5 ranacapa_0.1.0
# [17] devtools_2.4.2 usethis_2.1.5 microbiome_1.14.0 forcats_0.5.1
# [21] stringr_1.4.0 dplyr_1.0.7 purrr_0.3.4 readr_2.1.1
# [25] tidyr_1.1.4 tibble_3.1.6 ggplot2_3.3.5 tidyverse_1.3.1
# [29] phyloseq_1.36.0
#
# loaded via a namespace (and not attached):
# [1] readxl_1.3.1 backports_1.4.1 plyr_1.8.6 igraph_1.2.10
# [5] splines_4.1.0 BiocParallel_1.26.0 AlgDesign_1.2.0 digest_0.6.29
# [9] foreach_1.5.1 htmltools_0.5.2 fansi_0.4.2 magrittr_2.0.1
# [13] memoise_2.0.1 cluster_2.1.2 tzdb_0.2.0 remotes_2.4.2
# [17] annotate_1.70.0 Biostrings_2.60.0 modelr_0.1.8 prettyunits_1.1.1
# [21] colorspace_2.0-2 blob_1.2.2 rvest_1.0.2 haven_2.4.3
# [25] callr_3.7.0 crayon_1.4.2 RCurl_1.98-1.5 jsonlite_1.7.2
# [29] genefilter_1.74.0 survival_3.2-13 iterators_1.0.13 ape_5.6
# [33] glue_1.6.0 gtable_0.3.0 zlibbioc_1.38.0 XVector_0.32.0
# [37] DelayedArray_0.18.0 questionr_0.7.5 car_3.0-12 pkgbuild_1.3.1
# [41] Rhdf5lib_1.14.0 abind_1.4-5 scales_1.1.1 DBI_1.1.2
# [45] rstatix_0.7.0 miniUI_0.1.1.1 Rcpp_1.0.7 xtable_1.8-4
# [49] bit_4.0.4 httr_1.4.2 RColorBrewer_1.1-2 ellipsis_0.3.2
# [53] XML_3.99-0.8 pkgconfig_2.0.3 farver_2.1.0 dbplyr_2.1.1
# [57] locfit_1.5-9.4 utf8_1.2.2 AnnotationDbi_1.54.0 tidyselect_1.1.1
# [61] labeling_0.4.2 rlang_0.4.12 reshape2_1.4.4 later_1.2.0
# [65] munsell_0.5.0 cellranger_1.1.0 tools_4.1.0 cachem_1.0.6
# [69] cli_3.1.0 RSQLite_2.2.8 generics_0.1.1 ade4_1.7-18
# [73] broom_0.7.11 biomformat_1.20.0 fastmap_1.1.0 yaml_2.2.1
# [77] bit64_4.0.5 processx_3.5.2 fs_1.5.2 KEGGREST_1.32.0
# [81] nlme_3.1-153 mime_0.12 xml2_1.3.3 compiler_4.1.0
# [85] rstudioapi_0.13 png_0.1-7 curl_4.3.2 testthat_3.1.1
# [89] ggsignif_0.6.3 reprex_2.0.1 klaR_0.6-15 geneplotter_1.70.0
# [93] stringi_1.7.6 highr_0.9 ps_1.6.0 desc_1.4.0
# [97] Matrix_1.4-0 multtest_2.48.0 vctrs_0.3.8 pillar_1.6.4
# [101] lifecycle_1.0.1 rhdf5filters_1.4.0 combinat_0.0-8 data.table_1.14.0
# [105] bitops_1.0-7 httpuv_1.6.5 R6_2.5.1 promises_1.2.0.1
# [109] sessioninfo_1.2.2 codetools_0.2-18 MASS_7.3-54 assertthat_0.2.1
# [113] pkgload_1.2.4 rhdf5_2.36.0 rprojroot_2.0.2 withr_2.4.3
# [117] GenomeInfoDbData_1.2.7 mgcv_1.8-38 hms_1.1.1 grid_4.1.0
# [121] labelled_2.9.0 carData_3.0-5 Rtsne_0.15 shiny_1.7.1
# [125] lubridate_1.8.0
Please update the package from GitHub. I forgot to add na.rm = TRUE in the quantile() call.
Thanks for your reply @jokergoo
I removed the package using remove.packages
and I restarted R using .rs.restartR() every now and then
then
if (!requireNamespace("BiocManager", quietly=TRUE))
install.packages("BiocManager")
BiocManager::install("ComplexHeatmap")
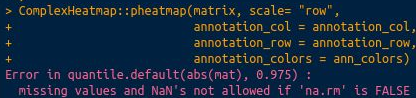
but it didn't work still giving the same error:
# Error in quantile.default(abs(mat), 0.975) :
# missing values and NaN's not allowed if 'na.rm' is FALSE
then I tried the dev version as follow and gave error as you can kindly see
remove.packages("ComplexHeatmap")
library(devtools)
install_github("jokergoo/ComplexHeatmap")
# Downloading GitHub repo jokergoo/ComplexHeatmap@HEAD
# ✓ checking for file ‘/tmp/RtmpdepRwf/remotes7b6b3f2e7dc2/jokergoo-ComplexHeatmap-ab225a0/DESCRIPTION’ ...
# ─ preparing ‘ComplexHeatmap’:
# ✓ checking DESCRIPTION meta-information ...
# ─ checking for LF line-endings in source and make files and shell scripts
# ─ checking for empty or unneeded directories
# ─ building ‘ComplexHeatmap_2.11.1.tar.gz’
#
# * installing *source* package ‘ComplexHeatmap’ ...
# ** using staged installation
# ** R
# Error in parse(outFile) :
# /tmp/RtmpBJSCmD/R.INSTALL809a76d9c93e/ComplexHeatmap/R/utils.R:103:56: unexpected symbol
# 111: message_wrap("The automatically generated colors map from the 1^st and 99^th of the values in the matrix. There are outliers in the matrix whose patterns might be h
# ^
# ERROR: unable to collate and parse R files for package ‘ComplexHeatmap’
# * removing ‘/home/r01mt19/.conda/envs/updatedR/lib/R/library/ComplexHeatmap’
# Warning message:
# In i.p(...) :
# installation of package ‘/tmp/RtmpdepRwf/file7b6b74f5e800/ComplexHeatmap_2.11.1.tar.gz’ had non-zero exit status
# > library(ComplexHeatmap)
# Error in library(ComplexHeatmap) :
# there is no package called ‘ComplexHeatmap’
sessionInfo()
# R version 4.1.0 (2021-05-18)
# Platform: x86_64-conda-linux-gnu (64-bit)
# Running under: Ubuntu 20.04.4 LTS
#
# Matrix products: default
# BLAS/LAPACK: /home/r01mt19/.conda/envs/updatedR/lib/libopenblasp-r0.3.18.so
#
# locale:
# [1] LC_CTYPE=en_GB.UTF-8 LC_NUMERIC=C LC_TIME=en_GB.UTF-8 LC_COLLATE=en_GB.UTF-8
# [5] LC_MONETARY=en_GB.UTF-8 LC_MESSAGES=en_GB.UTF-8 LC_PAPER=en_GB.UTF-8 LC_NAME=C
# [9] LC_ADDRESS=C LC_TELEPHONE=C LC_MEASUREMENT=en_GB.UTF-8 LC_IDENTIFICATION=C
#
# attached base packages:
# [1] stats graphics grDevices utils datasets methods base
#
# other attached packages:
# [1] devtools_2.4.2 usethis_2.1.5 phyloseq_1.36.0
#
# loaded via a namespace (and not attached):
# [1] nlme_3.1-153 bitops_1.0-7 matrixStats_0.61.0
# [4] fs_1.5.2 bit64_4.0.5 RColorBrewer_1.1-2
# [7] httr_1.4.2 rprojroot_2.0.2 GenomeInfoDb_1.28.0
# [10] tools_4.1.0 utf8_1.2.2 R6_2.5.1
# [13] vegan_2.5-7 DBI_1.1.2 BiocGenerics_0.40.0
# [16] mgcv_1.8-38 colorspace_2.0-3 permute_0.9-5
# [19] rhdf5filters_1.4.0 ade4_1.7-18 withr_2.4.3
# [22] prettyunits_1.1.1 processx_3.5.2 tidyselect_1.1.1
# [25] DESeq2_1.32.0 curl_4.3.2 bit_4.0.4
# [28] compiler_4.1.0 cli_3.1.0 Biobase_2.52.0
# [31] desc_1.4.0 DelayedArray_0.18.0 scales_1.1.1
# [34] genefilter_1.74.0 callr_3.7.0 stringr_1.4.0
# [37] XVector_0.32.0 pkgconfig_2.0.3 sessioninfo_1.2.2
# [40] MatrixGenerics_1.4.0 fastmap_1.1.0 rlang_0.4.12
# [43] rstudioapi_0.13 RSQLite_2.2.8 generics_0.1.1
# [46] jsonlite_1.7.2 BiocParallel_1.26.0 dplyr_1.0.7
# [49] RCurl_1.98-1.5 magrittr_2.0.1 GenomeInfoDbData_1.2.7
# [52] biomformat_1.20.0 Matrix_1.4-0 Rcpp_1.0.7
# [55] munsell_0.5.0 S4Vectors_0.32.3 Rhdf5lib_1.14.0
# [58] fansi_0.4.2 ape_5.6 lifecycle_1.0.1
# [61] stringi_1.7.6 yaml_2.2.1 MASS_7.3-54
# [64] SummarizedExperiment_1.22.0 zlibbioc_1.38.0 pkgbuild_1.3.1
# [67] rhdf5_2.36.0 plyr_1.8.6 grid_4.1.0
# [70] blob_1.2.2 parallel_4.1.0 crayon_1.5.0
# [73] lattice_0.20-45 Biostrings_2.60.0 splines_4.1.0
# [76] multtest_2.48.0 annotate_1.70.0 KEGGREST_1.32.0
# [79] locfit_1.5-9.4 ps_1.6.0 pillar_1.6.4
# [82] igraph_1.2.10 GenomicRanges_1.44.0 pkgload_1.2.4
# [85] geneplotter_1.70.0 reshape2_1.4.4 codetools_0.2-18
# [88] stats4_4.1.0 XML_3.99-0.8 glue_1.6.0
# [91] remotes_2.4.2 data.table_1.14.0 png_0.1-7
# [94] vctrs_0.3.8 foreach_1.5.2 testthat_3.1.1
# [97] gtable_0.3.0 purrr_0.3.4 assertthat_0.2.1
# [100] cachem_1.0.6 ggplot2_3.3.5 xtable_1.8-4
# [103] survival_3.2-13 tibble_3.1.6 iterators_1.0.14
# [106] AnnotationDbi_1.54.0 memoise_2.0.1 IRanges_2.28.0
# [109] cluster_2.1.2 ellipsis_0.3.2
You should directly update from GitHub:
library(devtools)
install_github("jokergoo/ComplexHeatmap")
And remember to restart your R session.
@jokergoo Many Thanks it worked now, it wasn't working before although I did restart and installed it directly from Github.
I am still getting an error which I can't solve although I tried different things: Error: number of observations in top annotation should be as same as ncol of the matrix.
ps.prev0
ps.prev0.zip
ps.prev.taxa <- tax_glom(ps.prev0, taxrank = "Species", NArm = FALSE)
ps.taxa.pse <- ps.prev.taxa
otu_table(ps.taxa.pse) <- otu_table(ps.prev.taxa) + 1
sample_data(ps.taxa.pse) # sanity check
tax_table(ps.taxa.pse)
# pairwise comparison between 2wk and 14wk post 1st feeding
deseq = phyloseq_to_deseq2(ps.taxa.pse, ~ dev.stage)
# converting counts to integer mode
# Warning message:
# In DESeqDataSet(se, design = design, ignoreRank) :
# some variables in design formula are characters, converting to factors
ds = DESeq(deseq, test="Wald", fitType="local")
# estimating size factors
# estimating dispersions
# gene-wise dispersion estimates
# mean-dispersion relationship
# final dispersion estimates
# fitting model and testing
# -- replacing outliers and refitting for 34 genes
# -- DESeq argument 'minReplicatesForReplace' = 7
# -- original counts are preserved in counts(dds)
# estimating dispersions
# fitting model and testing
alpha = 0.05
res = results(ds, alpha=alpha)
res = res[order(res$padj, na.last=NA), ]
taxa_sig = rownames(res[1:20, ]) # select bottom 20 with lowest p.adj values
ps.taxa.rel <- transform_sample_counts(ps.taxa.pse, function(x) x/sum(x)*100)
ps.taxa.rel.sig <- prune_taxa(taxa_sig, ps.taxa.rel)
matrix <- as.matrix(data.frame(otu_table(ps.taxa.rel.sig)))
colnames(matrix) <- as.character(tax_table(ps.taxa.rel.sig)[, "Species"])
metadata_sub <- data.frame(sample_data(ps.taxa.rel.sig))
# Define the annotation color for columns and rows
annotation_col = data.frame(
dev.stage = as.factor(metadata_sub$dev.stage),
check.names = FALSE
)
rownames(annotation_col) = rownames(metadata_sub)
annotation_row = data.frame(
Phylum = as.factor(tax_table(ps.taxa.rel.sig)[, "Phylum"]),
check.names = FALSE
)
rownames(annotation_row) = colnames(matrix)
# ann_color should be named vectors
phylum_col = RColorBrewer::brewer.pal(length(levels(annotation_row$Phylum)), "Paired")
names(phylum_col) = levels(annotation_row$Phylum)
ann_colors = list(
dev.stage = c(`2wk_Post1stFeeding` = "red", `14wk_Post1stFeeding` = "blue"),
Phylum = phylum_col
)
ComplexHeatmap::pheatmap(matrix, scale= "row",
annotation_col = annotation_col,
annotation_row = annotation_row_ed1,
annotation_colors = ann_colors)
Error: number of observations in top annotation should be as same as ncol of the matrix.
Can you tell me the values of the following variables?
dim(matrix)
dim(annotation_col)
dim(annotation_row_ed1)
Note nrow(matrix) should be the same as nrow(annotation_row_ed1), and ncol(matrix) should be the same as nrow(annotation_col).