Roman Chernyatchik
Roman Chernyatchik
 ``` rule a: input: "test.txt" log: "logs/a.log" conda: "envs/a.yaml" # TODO: syntax error in line 8 - ok, maybe we could better smth tell? output: "test.out" shell: "cat {input}...
Python option 'Smart indents on paste line' doesn't work properly ``` rule: input foo = "", output: "" ``` result: ``` rule: input foo = "", boo = ".."> output:...

E.g. we could use BOLD. ``` rule: input: "foo/{sample}.in" output: "foo/{sample}.out", "foo/{sample}.{config[genome]}.out", ``` Also this issue includes implementing Color Scheme page for Snakemake language
``` rule: output: "foo/{sample}.{config[genome]}.out", ``` Here better highlight whole `config[genome]` as wildcard
A warning that warns if some section name couldn't be used with project snakemake version (from settings), subtask of #334
At the moment parser inserts `Py:TRIPPLE_QUOTED_STRING` instead of `Py:DOCSTRING`, see screenshot. So we get wrong highlighting.  ``` rule foo: """ doc0 """ log: "foo.log" """ doc1 """ output: touch("foo.out")...
Add warning which detects misspelled section names, e.g we could use Levenshtein or etc distance to predict the expected section name + quick fix, subtask of #334
Not always work, e.g on update: ``` 2021-04-24 17:12:00,837 [ 79] INFO - #com.intellij.idea.Main - JVM: 11.0.10+9-b1341.41 (Dynamic Code Evolution 64-Bit Server VM) 2021-04-24 21:13:19,074 [14478316] INFO - lij.ide.plugins.DynamicPlugins -...
Duplicated inspection errors: 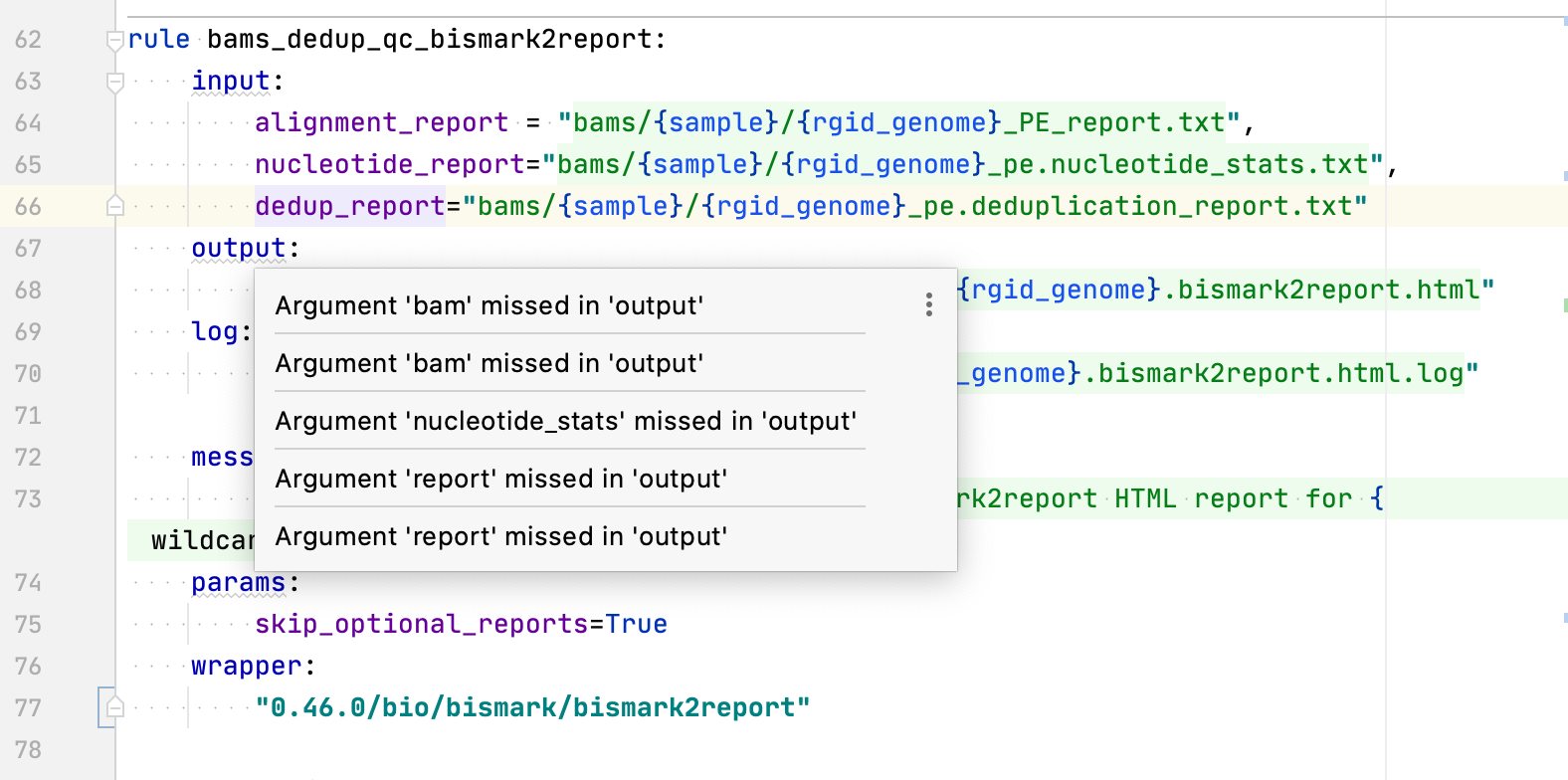 ``` rule bams_dedup_qc_bismark2report: input: alignment_report = "bams/{sample}/{rgid_genome}_PE_report.txt", nucleotide_report="bams/{sample}/{rgid_genome}_pe.nucleotide_stats.txt", dedup_report="bams/{sample}/{rgid_genome}_pe.deduplication_report.txt" output: html="qc/bams_dedup/bismark2report/{sample}/{rgid_genome}.bismark2report.html" log: "qc/bams_dedup/bismark2report/{sample}/{rgid_genome}.bismark2report.html.log" message: "QC: Deduplicated [{wildcards.sample}] bismark2report HTML report for {wildcards.rgid_genome}" params: skip_optional_reports=True wrapper: "0.46.0/bio/bismark/bismark2report"...
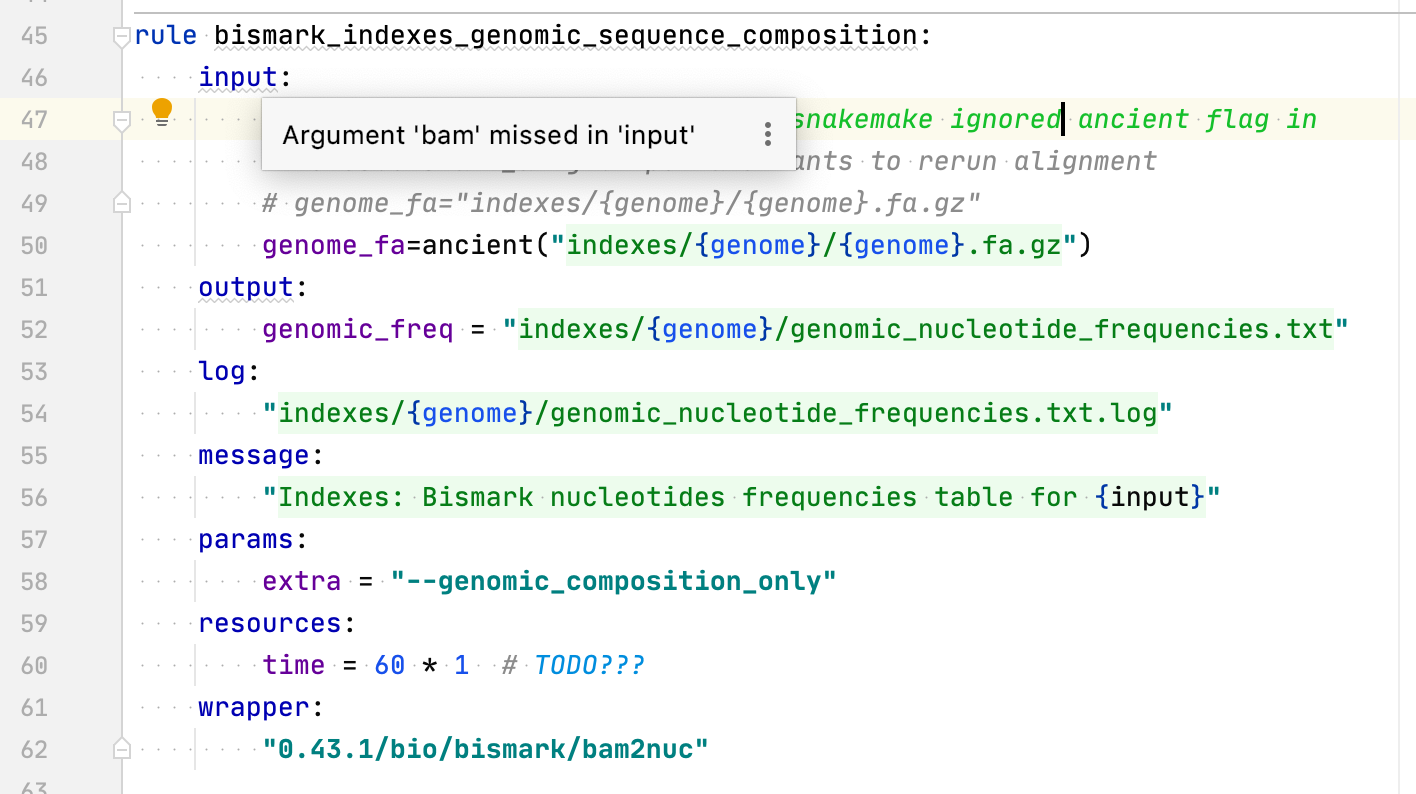 See https://snakemake-wrappers.readthedocs.io/en/0.64.0/wrappers/bismark/bam2nuc.html ``` bam = snakemake.input.get("bam", None) if bam: cmdline_args.append("{bam}") ... else: cmdline_args.append("--genomic_composition_only") ```