openff-toolkit
 openff-toolkit copied to clipboard
openff-toolkit copied to clipboard
Unassigned angle/torsion parameter
Describe the bug I was looking into the Gen3 torsions set, and there seems to be some chemical perception issue with Nitrogens in resonance cases, or "[#7X3+1]=" type. Here is a notebook with 59 such molecules where some pass through but some fail angle parameter assignment, or torsion parameter assignment.
Charge assignment failures: 0 Angle assignment failures: 1 Proper torsion assignment failures: 13 Total failures: 14 out of 59
To Reproduce Notebook here: nitrogen_failures.tar.gz Pdf of notebook for easy molecule viewing: check_nitrogen_parameters.pdf
Making a note that new parameters with the smarts patterns "[#7X3+1]-[#6X4]" , "[#7X3+1]-[#7X3]" for central bonds in torsions, and "[#8X]=,:[#7X3+1]-[*]" for angle may cover these, until further discussion.
Is this a new issue (finding some molecules that should be assigned parameters, but aren't) or a regression (an old version of the toolkit did assign parameters, but recent versions aren't)?
I think it is a new issue, mostly lack of parameters and not toolkit related.
Wherever is best for you and the fitting team to keep track of reports of missing parameters - if that's here that's not a problem. I just wanted to determine if this was a bug in the code or not.
Just making a note for some failures, since these may not be important chemistries we can try to modify the general parameters to catch them
- for molecules of type (failure for central bond [3,5])
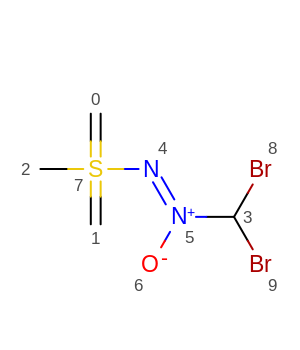 simple solution would be to make
simple solution would be to make t51: [*:1]-[#6X4:2]-[#7X3:3]-[*:4]"more general with the first and third bonds being more flexible[*:1]~[#6X4:2]-[#7X3:3]~[*:4]. - for molecules of type (failure for central bond [8,6])
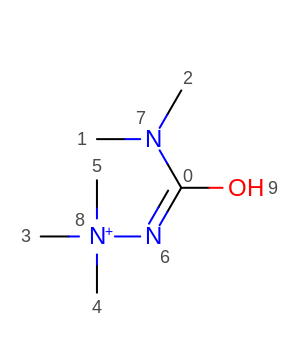
t138: "[*:1]~[#7X2:2]-[#7X3:3]~[*:4]"to include#7X4as well in the third position, making it"[*:1]~[#7X2:2]-[#7X3,#7X4:3]~[*:4]"