f5c
 f5c copied to clipboard
f5c copied to clipboard
avoid skip when using f5c eventalign
Hi f5C team! I wonder if there are any settings to avoid event skip when using f5c eventalign? I found not all bases in reference would have event in f5c eventalign results.
At the moment it does not expose an option. But I can see if this is something that I can quickly expose as an option. Could you please give a bit more information and probably an example so that I can better understand the context?
Hi! Thank you so much for your reply! Below picture shows a example of skip (5293 and 5294 not found). I want to develop methods to detect methylation which need to look into raw signal assigned to the query bases, but if skip occurs in this base, I have to give up this read. Unfortunately, there many skips in the eventalign results.

There are a couple of cases where this kind of skip occurs.
- Actual deletions where it is not possible to have corresponding signal points that relate to those deleted bases in the reference genome
- The event segmentation algorithm does undersegmenting which then propagates as a skip.
Is the above example a real deletion (you can the corresponding position in IGV something)?
Hi! There is not a real deletion. I can find many skips like below. I wonder if there are any settings to avoid event skip, as you know when sequencing, each base has its event.

Currently, eventalign does not have an option. But I can see if I could implement it - need to understand the context clearly first for that.
Eventalign gives the output against the reference genome. Is that what you are after or do you want the alignment against the basecalled read?
Hi! What I want to do is to assign events to each base in the reference genome, but I found many bases do not have its event (be skipped).
unfortunately, around 1% of bases can be skips. While it could be possible to reduce this (which needs some implementation work), it is not possible to achieve 0% due to the noise in both the time and amplitude axis in the raw signal. What is the percentage of skips for your dataset?
For instance, in the example below, see the marked area where one base/k-mer is missing (the crossed one),
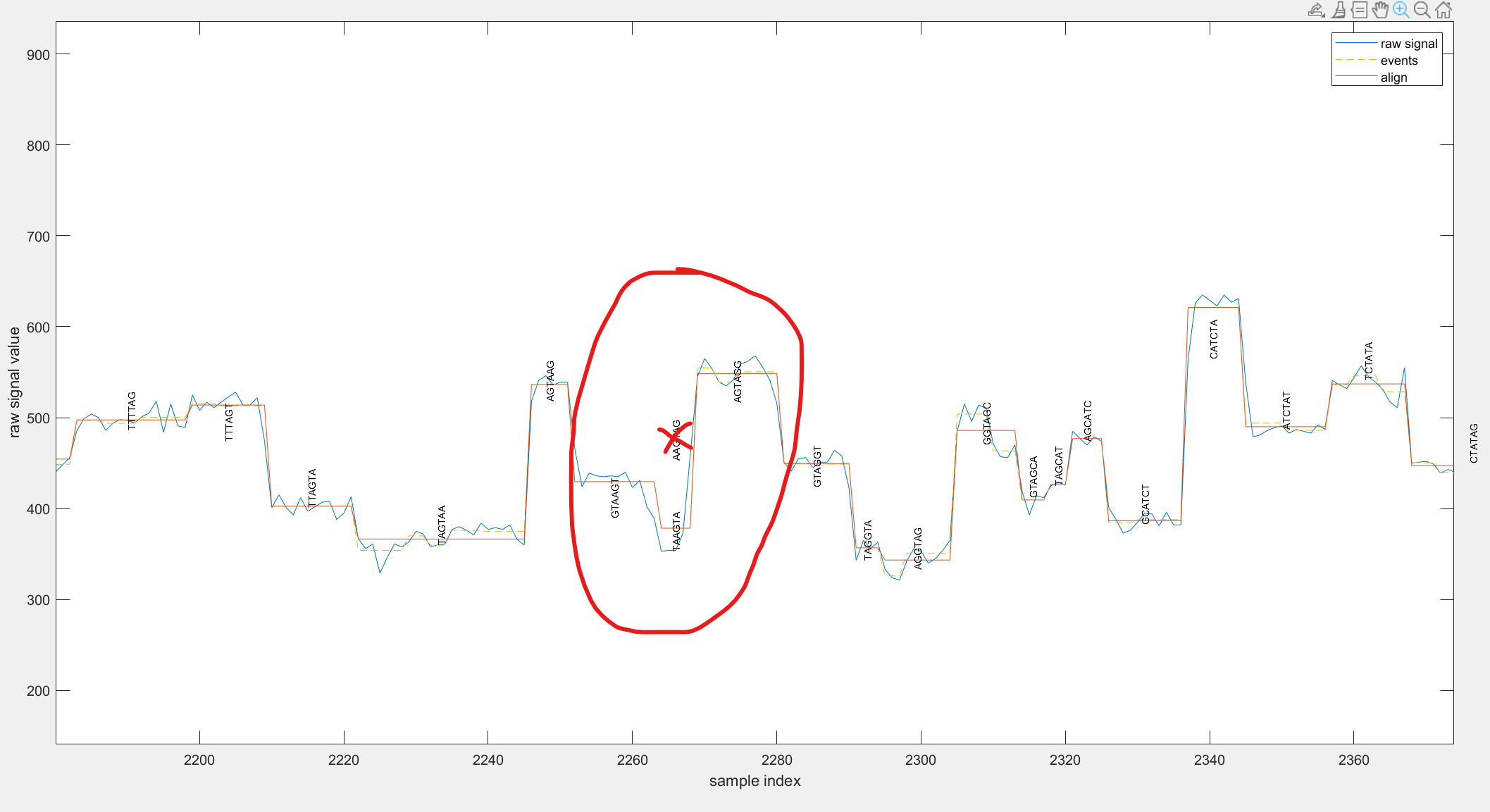
As you can see around that area, the amount of variation in the signal is not that much and the event segmentation has failed to split it into two events. We can force it to be split, but I am not sure if it will give anything beneficial in the downstream analysis. I think the best strategy is to ignore such bases in the downstream analysis thinking of this ignoring as kind of a filter for low quality areas.