Question about: RuntimeWarning: invalid value encountered in log
Hi,
When running xpclr with TXT files, i got a problem.
` 2020-06-10 23:45:20 : INFO : running xpclr v1.1.2
2020-06-10 23:45:20 : INFO : Loading TXT
2020-06-10 23:50:12 : INFO : TXT loading complete
2020-06-10 23:50:15 : INFO : 146,862 SNPs in total are in the provided input files
2020-06-10 23:50:15 : INFO : 0 SNPs excluded as multiallelic
2020-06-10 23:50:15 : INFO : 0 SNPs excluded as missing in all samples in a population
2020-06-10 23:50:15 : INFO : 7,813 SNPs excluded as invariant or singleton in population 2
2020-06-10 23:50:15 : INFO : 139,049/146,862 SNPs included in the analysis (94.68%)
2020-06-10 23:50:17 : INFO : Done dropping above SNPs from analysis. XP-CLR algorithm starting.
2020-06-10 23:50:21 : INFO : Omega estimated as : 0.573789
~/anaconda3/lib/python3.7/site-packages/xpclr/methods.py:146: RuntimeWarning: invalid value encountered in log ratio = np.log(like_i) - np.log(like_b)
2020-06-11 05:26:53 : INFO : Analysis complete. Output file ~/xpclr_python/out_25k `
command-line is 'xpclr --out ~/xpclr_python/out_25k --format txt --map ~/xpclr/snp_pos --popA ~/xpclr/file_A --popB ~/xpclr/file_B --chr 25 --phased --maxsnps 600 --size 25000 --step 10000'
I wonder how this RuntimeWarning changed the result of output, because i check the output is OK. I use other chromsomes with no RuntimeWarning, so i think it is relevant to the input file, but i don't know the reason.
Hmm- what seems to be happening is that a likelihood is below 0 before the np.log is applied.
This might be an edge case- where the area under the curve is tiny and a rounding error causes the value to go below 0, or possibly a bug. If you can share the output file, or at least the row that has the NaN likelihood ratio, I can try to debug.
Hmm- what seems to be happening is that a likelihood is below 0 before the
np.logis applied.This might be an edge case- where the area under the curve is tiny and a rounding error causes the value to go below 0, or possibly a bug. If you can share the output file, or at least the row that has the NaN likelihood ratio, I can try to debug.
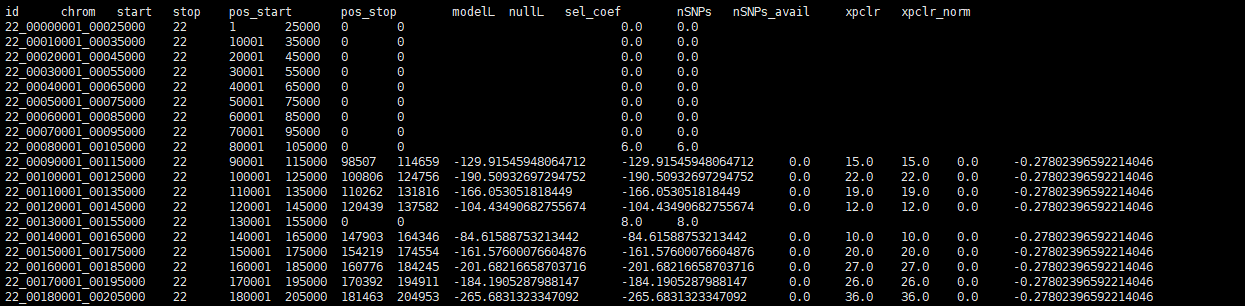
I am sorry, i can't position which row is the question row because the error info didn't show it. I check the result, the row number is correct. I am not sure if the row like the photo below is the question row, because i can also find these rows in the no-warning result.
in RuntimeWarning result:
in no-warning result:
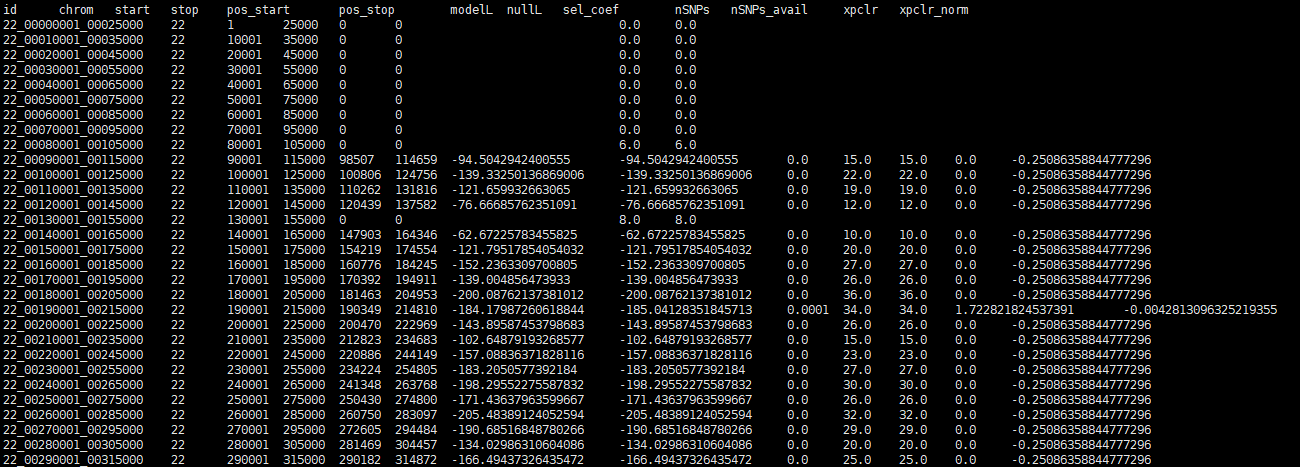
Thanks. In the RunTime Warning output- do any rows have a Nan value for the xpclr column? If so, can you share those rows?
Thanks. In the
RunTime Warningoutput- do any rows have aNanvalue for thexpclrcolumn? If so, can you share those rows?
I checked the RunTime Warning output, all the rows with 'NaN' 'xpclr' column are like what i marked below. 'pos_start','pos_stop' value is '0', 'modelL','nullL','sel_coef','xpclr','xpclr_norm' value is 'NaN' , ‘nSNPs’,‘nSNPs_avail’ value is normal.
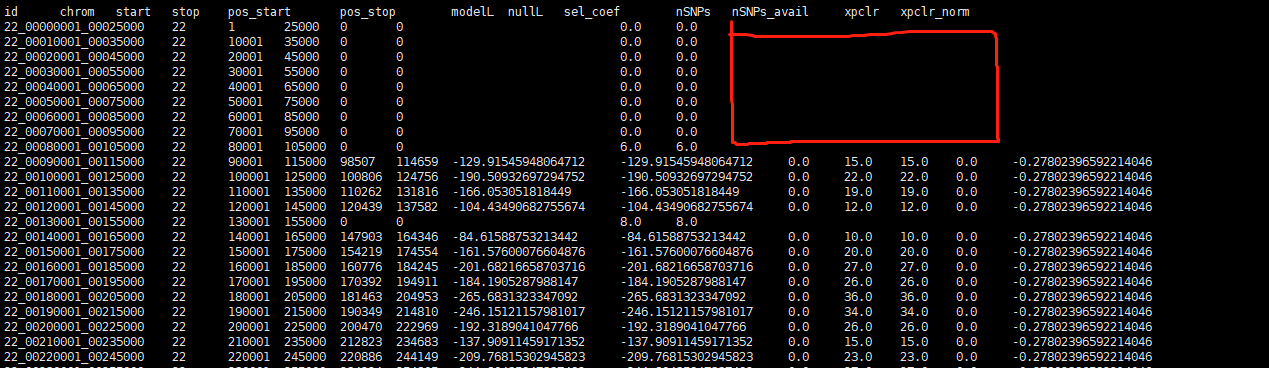
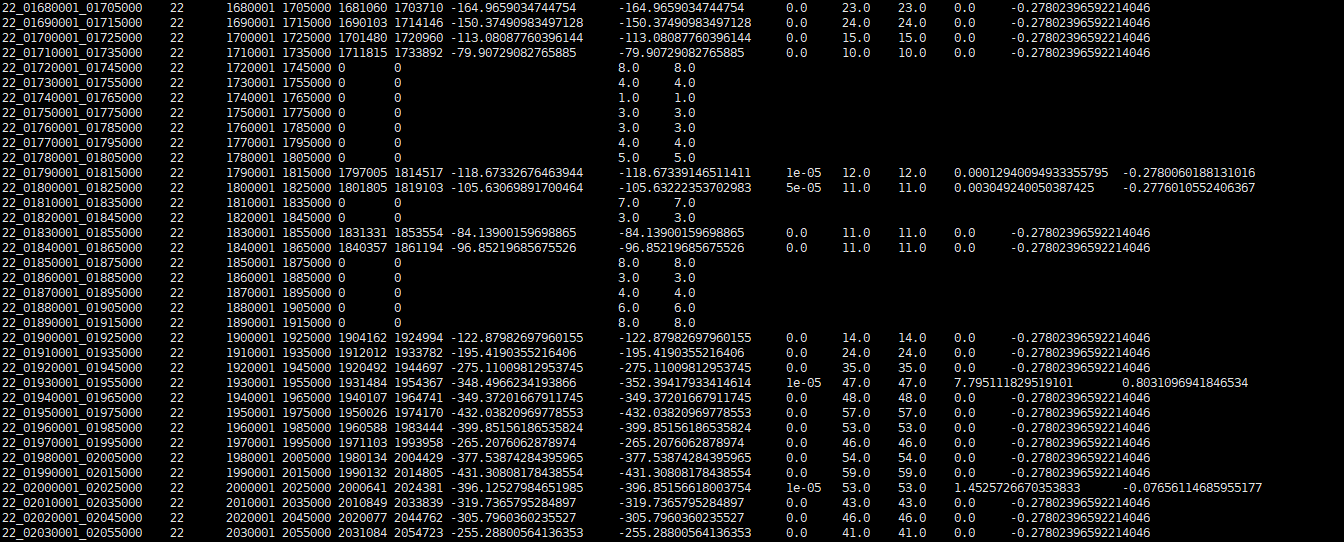
I think that's a slightly different issue. Where there are no SNPs, your likelihoods should be 0, which when you log is -Inf. Which gives a distinct warning.
The warning here suggests that your likelihoods are negative for some reason.
I should add a check for negative likelihoods- I'll leave this open to do so in the next version.
For now though, I think it shouldn't change your conclusions, I think it's most likely an issue with rounding.
Generally though, it looks like your windows are too small. I would suggest increasing them so you include ~200 SNPs in each. Given you have ~50 now, I think 100_000 would be a reasonable size.
I think that's a slightly different issue. Where there are no SNPs, your likelihoods should be 0, which when you log is
-Inf. Which gives a distinct warning. The warning here suggests that your likelihoods are negative for some reason.I should add a check for negative likelihoods- I'll leave this open to do so in the next version.
For now though, I think it shouldn't change your conclusions, I think it's most likely an issue with rounding.
Generally though, it looks like your windows are too small. I would suggest increasing them so you include ~200 SNPs in each. Given you have ~50 now, I think
100_000would be a reasonable size.
I will increase the window size, thanks for your help.