chromap
 chromap copied to clipboard
chromap copied to clipboard
Is there a proper way to generate bigwig file from bed output?
I need to use deeptools to analysis my bulk ATAC-seq data. However, deeptools require a bigwig (.bw) file for most of the analyses. I have tried two pipelines to generate bigwig file.
Pipeline 1:
- Generate a bed file by chromap
- Generate a bedgraph file from bed file with 'bedtools genomecov -bg'
- Sort the bedgraph file with 'bedtools sort'
- Convert the bedgraph file to a bigwig file by 'bedGraphToBigWig' from http://hgdownload.soe.ucsc.edu/admin/exe/
- TSS enrichment analysis with 'computeMatrix' and 'plotHeatmap' from deeptools
Pipeline 2:
- Generate a sam file by chromap
- Convert the sam file to a bam file by 'samtools view -S -b'
- Generate a bigwig file by 'bamCoverage' from deeptools.
- TSS enrichment analysis with 'computeMatrix' and 'plotHeatmap' from deeptools
However, the final heatmap from these two pipelines were completely different. The heatmap from Pipeline 1 was completely black.
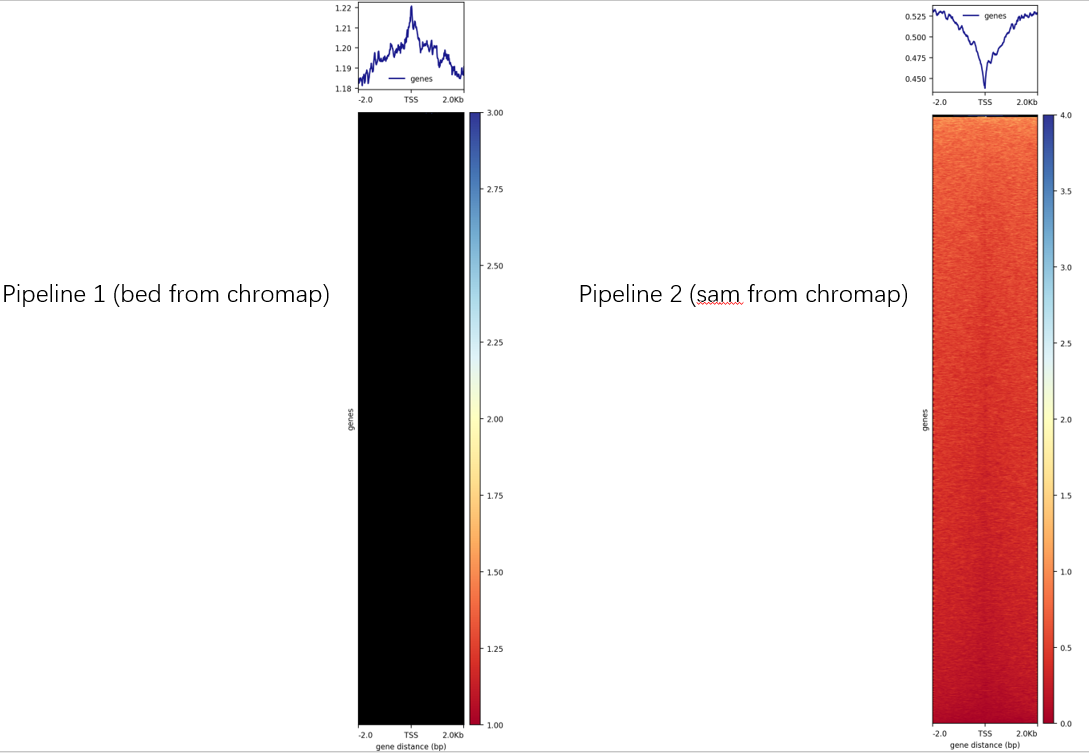
I provide the two .bw files from these two pipelines at https://github.com/loraince/miscellaneous/tree/main/bigwig_test When visualizing these two .bw files in IGV, they have slight differences in details, which is not a big problem.
The result from Pipeline 2 have a similar heatmap as the result from a common bwa -> deeptools pipelines. So I guess there is a problem with Pipeline 1. I wonder if there is a better way to generate bigwig file with bed file from chromap ?
The parameters of chromap are as follows for the two pipelines: chromap --preset atac -x {index} -r {fasta} -t {threads} -1 {input.R1} -2 {input.R2} --SAM -o {output.sam} chromap --preset atac -x {index} -r {fasta} -t {threads} -1 {input.R1} -2 {input.R2} -o {output.bed}
By the way, don't mind the strange pit of the TSS enrichment analysis,. We are testing some special technique. You can try these two pipelines with any handy bulk ATAC-seq data.
It seems that deeptools only support SAM/BAM input. One possible solution is to convert the bed files to bam files using bedtools. You may have to add the header to the bam using samtools if that is required by deeptools. And then run the rest of the pipeline 2. But it would still be much better to come up with a pipeline to directly get the bigwig file from bed. I guess 'bamCoverage' is doing normalization or other processing when generating bigwig file, which makes its output value ranges different from your resulting value ranges in the bigwig file in pipeline 1.
Can you also start an issue in the deeptools repo and ask them the recommended way to use bed as an input for the analysis?
It seems that deeptools only support SAM/BAM input. One possible solution is to convert the bed files to bam files using bedtools. You may have to add the header to the bam using samtools if that is required by deeptools. And then run the rest of the pipeline 2. But it would still be much better to come up with a pipeline to directly get the bigwig file from bed. I guess 'bamCoverage' is doing normalization or other processing when generating bigwig file, which makes its output value ranges different from your resulting value ranges in the bigwig file in pipeline 1.
Can you also start an issue in the deeptools repo and ask them the recommended way to use bed as an input for the analysis?
Thank you very much. I tried the bedToBam from bedtools and it worked well with the subsequent steps in pipeline 2.
@loraince thanks for sharing this. I too am going to use deepTools2 for a ChIP-seq experiment (not ATAC) and I wanted to confirm that your bed output from chromap converted back to a bam with bedtools gives you the same result as just outputing a sam file from chromap? Just wanted to make sure that the bedtools conversion back to a bam works ok with deepTools (and gets the results expected). If so, I will borrow this workflow as well.
Thanks!!
@loraince @haowenz Dear Loraince. I'm doing similar work as yours but I can't generate bigwig file successfully. Can you please share the commands you used after getting the bed file from Chromap? What I did is just chromap --preset chip -x {index} -r {fasta} -t {threads} -1 {input.R1} -2 {input.R2} -o {output.bed} bedToBam -i {chromap_output.bed} -g lower_TAIR10.size.genome > chromap.bam samtools index -b sorted_CRR303825_chromap.bam bamCoverage -bs 20 --effectiveGenomeSize 119481543 -b chromap.bam -of bigwig -o chromap.bigwig but the bamCoverage step always failed. Did I miss something important? Looking forward to your reply!