chromap
 chromap copied to clipboard
chromap copied to clipboard
Weird length of mapped fragments of ATAC-seq data
Chromap is indeed an ultrafast tool. Thanks for developing and this method. However, I have encountered two problems when I using chromap with scATAC-seq data.
The first one is that when I analyzed the distribution of fragment length, I found a dramatic loss of fragments between 47bp-120bp
The results are shown in these figures.
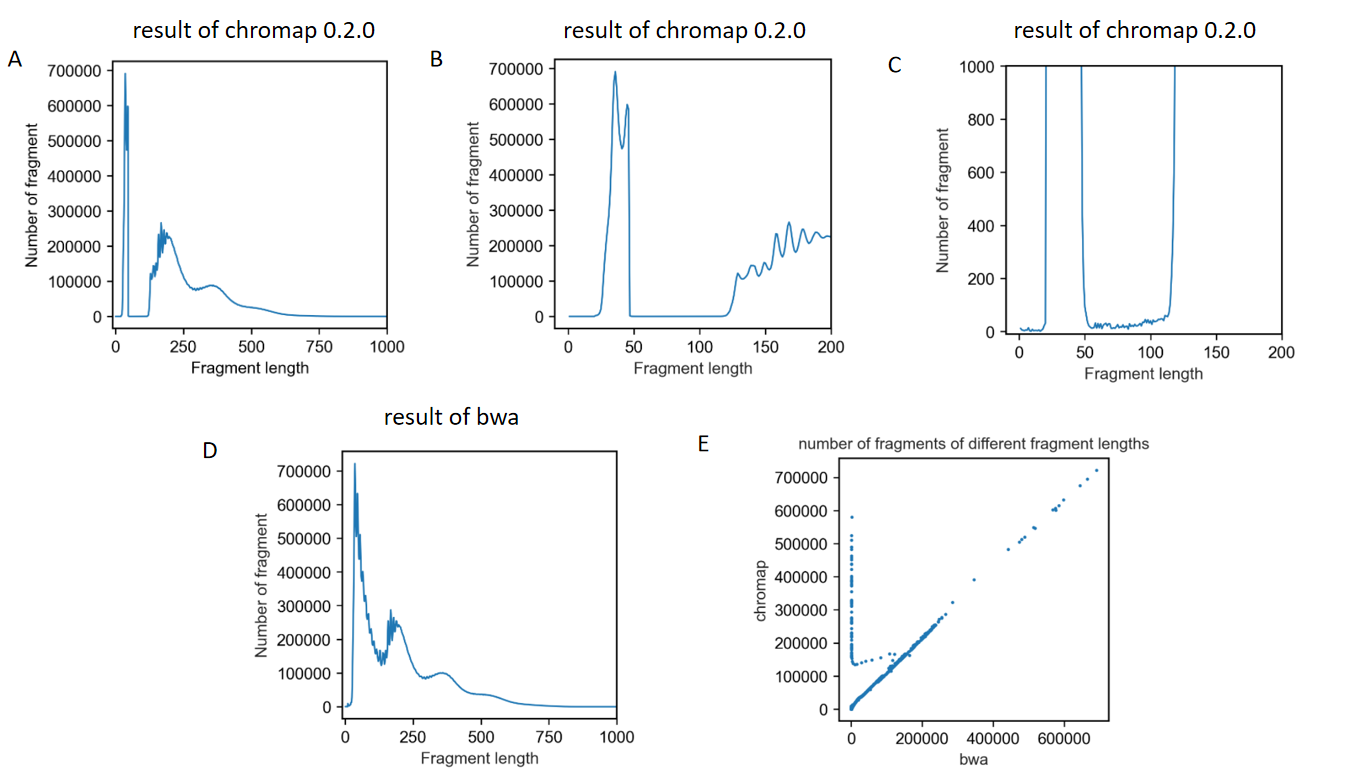
Another problem is that fragments of 0bp existed in the result bed file.

These two problems existed for all data of my different experiments. All these data have 10x-like scATAC-seq structures, using 150bp paired end sequencing.
The command was like this:
chromap --preset atac -x ${index} -r ${fasta} -1 ${inputR1} -2 ${inputR3} -o ${output} -b ${inputR2} --barcode-whitelist ${whitelist}
I also tried without the barcode fastq, running like this: chromap --preset atac -x ${index} -r ${fasta} -1 ${inputR1} -2 ${inputR3} -o ${output}
However, the two problems still existed for my data.
When I tried another scATAC-seq dataset with 50bp paired end sequencing, there two problems did not appear.
Thanks for sharing your test results with us. Indeed, the length distribution change is too sharp, and the fragment length 0 should not happen either. Is the data publicly available? If it is not, could you please share a few hundreds of the paired-end sequence data with us (no need for the barcode file)? We will look into this issue. Thank you.
Thank you for your reply.
I uploaded 100,000 read pairs of my paired fastq here without barcode file: https://github.com/loraince/miscellaneous/blob/main/ATAC_100k.zip
This data contains both human cells and mouse cells. When mapping to GRCh38, one fragment with length 0 appeared at chr5 134927308 134927308.
Here are the distribution of fragment length.
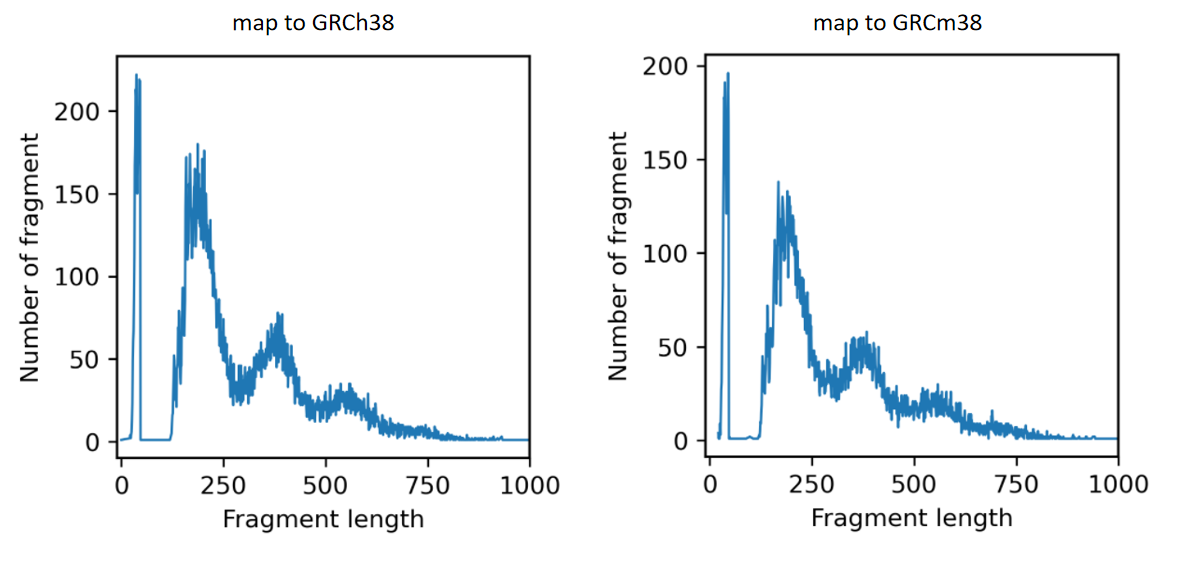
It is worth noting that ATAC_100k_1.fq has 150 bp for each read, while ATAC_100k_2.fq has 55 bp for each read. That is because the barcode is a part of the original R2 file and I split it to get the barcode file and a truncated ATAC_100k_2.fq. I guess the unequal length of R1.fq and R2.fq may cause the problem.
Thank you for the updation. I tried with the v0.2.2 and the distribution of fragment length is correct now.
However, the fragments with length 0 still exist. For example, when mapping the small dataset above to the human genome, one fragment was recorded with 0 bp at chr5 134927308 134927308. I am not sure whether you can reproduce this. If not, maybe the difference is between the reference genome. I am unsing human genome from http://cistrome.org/~galib/MAESTRO/references/scATAC/Refdata_scATAC_MAESTRO_GRCh38_1.1.0.tar.gz
By the way, could you please help me with two other questions?
- I find that --trim-adapters is true in --preset atac, while it is not used in --preset chip. I wonder why not trim adapters by default when it comes to chip-seq mode.
- Sometimes we need a BAM file for the downstream analysis. Is there any way to output a BAM file?
Thank you for the updation. I tried with the v0.2.2 and the distribution of fragment length is correct now.
However, the fragments with length 0 still exist. For example, when mapping the small dataset above to the human genome, one fragment was recorded with 0 bp at chr5 134927308 134927308. I am not sure whether you can reproduce this. If not, maybe the difference is between the reference genome. I am unsing human genome from http://cistrome.org/~galib/MAESTRO/references/scATAC/Refdata_scATAC_MAESTRO_GRCh38_1.1.0.tar.gz
Thank you for testing it again. We will check what happened.
By the way, could you please help me with two other questions?
- I find that --trim-adapters is true in --preset atac, while it is not used in --preset chip. I wonder why not trim adapters by default when it comes to chip-seq mode.
The adapter trimming we have, if you had a look at the paper, since we don't require user to input the adapter sequences, it only trims when are adapters on 3' end. In this case, the two ends of the read pair have an overlap in the middle which is the true fragment (see the last supplementary figure). Our method can detect this overlap and trim the adapters on 3' end. Note that this case happens a lot for scATAC-seq since the fragment length of scATAC-seq can frequently be short like 30-50bp (shorter than read length). On the other hand, for ChIP-seq, the fragment length is usually long. So we don't turn it on by default. But you can turn it on and see how much difference you would observe. In most cases, the difference on ChIP-seq should be very minor.
- Sometimes we need a BAM file for the downstream analysis. Is there any way to output a BAM file?
Currently, Chromap cannot output BAM directly. One way to get BAM is to output mappings in SAM and then convert it to BAM using samtools, just like the way you get BAM when using other aligners (e.g., BWA-MEM). The initial goal of designing Chromap is to bypass SAM and generate fragment files directly, which would save a lot of time. That's why we haven't put much effort on generating SAM or even BAM. Well, we do plan to add BAM support. But it would require a lot of works and it won't happen very soon in the future.
Hi @loraince, the fragment length 0 alignments seem to be chimeric reads or other sequencing artifacts. The example you mention should be the read: A00253:575:HWWVTDSX2:4:1260:28691:24314, where BWA-MEM aligned it on chrM and also creates a very short fragment size (9). It is by chance that Chromap selects another alignment coordinate with fragment 0. The downstream analysis tool usually filters such short fragments, so I don't think you need to worry about the case.
Thank you both so much for such prompt and detailed replies. My questions have been sufficiently addressed.