metacoder
 metacoder copied to clipboard
metacoder copied to clipboard
otu_table, tax_data and sample_data only contains NA after parsing from phyloseq
Hi,
I am trying to parse my phyloseq object to a taxmap object but I cant get it to work properly. The parsing seems to work fine (No error message) but when I look at the taxmap object it looks like this:
1284 taxa: aab. Bacteria ... bxk. TACGTAGGGGGCGA[truncated] 1284 edges: NA->aab, NA->aac, aab->aad, aab->aae, aab->aaf ... anv->bxh, anu->bxi, axg->bxj, anu->bxk 4 data sets: otu_table:
NA NA NANANA NANANA NANANA tax_data:
NA NA NANANA NANANA NANANA sample_data:
NA NA phy_tree:
Phylogenetic tree with 680 tips and 678 internal nodes. Tip labels: ASV1, ASV2, ASV3, ASV4, ASV5, ASV6, ... Node labels: , 0.872, 0.889, 0.000, 0.737, 0.857, ... Unrooted; includes branch lengths.0 functions:
Any idea whats causing this and how to resolve it?
Hi @FFoQS90,
That error looks familiar, but I cant remember what causes it. Can you send my your phyloseq object (or another that causes the same problem) using the save function in R? You can email me at zacharyfoster1989 gmail.com if you don't want to attach it here. Thanks
Sorry for the delay. I cant reproduce that problem:
library(metacoder)
#> Loading required package: taxa
#> This is metacoder verison 0.3.3 (stable)
x = readRDS('~/Downloads/pobj.rds')
parse_phyloseq(x)
#> Loading required package: phyloseq
#>
#> Attaching package: 'phyloseq'
#> The following object is masked from 'package:taxa':
#>
#> filter_taxa
#> <Taxmap>
#> 1284 taxa: aab. Bacteria ... bxk. TACGTAGGGGGCGA[truncated]
#> 1284 edges: NA->aab, NA->aac ... axg->bxj, anu->bxk
#> 4 data sets:
#> otu_table:
#> # A tibble: 680 x 52
#> taxon_id otu_id `1` `10` `11` `12` `13` `14` `15`
#> <chr> <chr> <int> <int> <int> <int> <int> <int> <int>
#> 1 axh ASV1 23 19 28 40 20 77 94
#> 2 axi ASV2 5 4 1 2 0 17 2
#> 3 axj ASV3 5 6 0 19 5 6 18
#> # … with 677 more rows, and 43 more variables: `16` <int>,
#> # `17` <int>, `18` <int>, `19` <int>, `2` <int>,
#> # `20` <int>, `21` <int>, `22` <int>, `23` <int>,
#> # `24` <int>, …
#> tax_data:
#> # A tibble: 680 x 10
#> taxon_id otu_id Kingdom Phylum Class Order Family Genus
#> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr>
#> 1 axh ASV1 Bacter… Firmi… Baci… Baci… Bacil… Virg…
#> 2 axi ASV2 Bacter… Firmi… Baci… Baci… Bacil… Virg…
#> 3 axj ASV3 Bacter… Firmi… Baci… Baci… Bacil… Orni…
#> # … with 677 more rows, and 2 more variables:
#> # Species <chr>, ASV <chr>
#> sample_data:
#> # A tibble: 50 x 28
#> sample_id Sample.ID Treatment Days Strain Combo Moisture
#> <chr> <chr> <chr> <chr> <chr> <chr> <chr>
#> 1 1 1 PLE 0 SH.28… PLE.… 0.182
#> 2 10 10 PLE 14 SH.28… PLE.… 0.142
#> 3 11 11 PLE 14 SH.28… PLE.… 0.165
#> # … with 47 more rows, and 21 more variables:
#> # Salmonella <chr>, pH <chr>, Aw <chr>, TOC <chr>,
#> # TC <chr>, IC <chr>, TN <chr>, NH4 <chr>, NO3 <chr>,
#> # Al <chr>, …
#> phy_tree:
#>
#> Phylogenetic tree with 680 tips and 678 internal nodes.
#>
#> Tip labels:
#> ASV1, ASV2, ASV3, ASV4, ASV5, ASV6, ...
#> Node labels:
#> , 0.872, 0.889, 0.000, 0.737, 0.857, ...
#>
#> Unrooted; includes branch lengths.
#> 0 functions:
Created on 2019-10-15 by the reprex package (v0.3.0)
Can you reinstall metacoder and phyloseq and try again? Also, your sessionInfo()might be useful. Thanks!
Thank you for looking into this. I reinstalled phyloseq and metacoder still the same problem. Here is my sessionInfo:
> sessionInfo()
R version 3.6.1 (2019-07-05)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 18.04.3 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.7.1
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.7.1
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=de_AT.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=de_AT.UTF-8 LC_MESSAGES=en_US.UTF-8 LC_PAPER=de_AT.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C LC_MEASUREMENT=de_AT.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] metacoder_0.3.3 taxa_0.3.2 phyloseq_1.28.0 dada2_1.12.1 Rcpp_1.0.2
loaded via a namespace (and not attached):
[1] colorspace_1.4-1 hwriter_1.3.2 htmlTable_1.13.2 XVector_0.24.0
[5] GenomicRanges_1.36.1 base64enc_0.1-3 rstudioapi_0.10 farver_1.1.0
[9] remotes_2.1.0 ggrepel_0.8.1 ggnet_0.1.0 bit64_0.9-7
[13] AnnotationDbi_1.46.1 lubridate_1.7.4 codetools_0.2-16 splines_3.6.1
[17] doParallel_1.0.15 geneplotter_1.62.0 knitr_1.25 polyclip_1.10-0
[21] zeallot_0.1.0 ade4_1.7-13 Formula_1.2-3 jsonlite_1.6
[25] Rsamtools_2.0.3 annotate_1.62.0 cluster_2.1.0 ggforce_0.3.1
[29] compiler_3.6.1 httr_1.4.1 backports_1.1.5 assertthat_0.2.1
[33] Matrix_1.2-17 lazyeval_0.2.2 tsnemicrobiota_0.1.0 cli_1.1.0
[37] tweenr_1.0.1 acepack_1.4.1 htmltools_0.4.0 tools_3.6.1
[41] igraph_1.2.4.1 gtable_0.3.0 glue_1.3.1 GenomeInfoDbData_1.2.1
[45] reshape2_1.4.3 dplyr_0.8.3 ShortRead_1.42.0 Biobase_2.44.0
[49] vctrs_0.2.0 Biostrings_2.52.0 multtest_2.40.0 ape_5.3
[53] nlme_3.1-141 iterators_1.0.12 xfun_0.10 stringr_1.4.0
[57] network_1.15 lifecycle_0.1.0 XML_3.98-1.20 zlibbioc_1.30.0
[61] MASS_7.3-51.4 scales_1.0.0 parallel_3.6.1 SummarizedExperiment_1.14.1
[65] biomformat_1.12.0 rhdf5_2.28.1 RColorBrewer_1.1-2 memoise_1.1.0
[69] gridExtra_2.3 ggplot2_3.2.1 rpart_4.1-15 latticeExtra_0.6-28
[73] stringi_1.4.3 RSQLite_2.1.2 genefilter_1.66.0 S4Vectors_0.22.1
[77] foreach_1.4.7 checkmate_1.9.4 permute_0.9-5 BiocGenerics_0.30.0
[81] BiocParallel_1.18.1 GenomeInfoDb_1.20.0 rlang_0.4.0 pkgconfig_2.0.3
[85] matrixStats_0.55.0 bitops_1.0-6 lattice_0.20-38 purrr_0.3.2
[89] Rhdf5lib_1.6.2 GenomicAlignments_1.20.1 htmlwidgets_1.5.1 cowplot_1.0.0
[93] bit_1.1-14 tidyselect_0.2.5 plyr_1.8.4 magrittr_1.5
[97] DESeq2_1.24.0 R6_2.4.0 IRanges_2.18.3 Hmisc_4.2-0
[101] DelayedArray_0.10.0 DBI_1.0.0 pillar_1.4.2 foreign_0.8-72
[105] mgcv_1.8-29 survival_2.44-1.1 RCurl_1.95-4.12 nnet_7.3-12
[109] tibble_2.1.3 crayon_1.3.4 plotly_4.9.0 locfit_1.5-9.1
[113] grid_3.6.1 data.table_1.12.4 blob_1.2.0 vegan_2.5-6
[117] digest_0.6.21 xtable_1.8-4 tidyr_1.0.0 RcppParallel_4.4.4
[121] stats4_3.6.1 munsell_0.5.0 viridisLite_0.3.0 ampvis2_2.5.1
Thanks!
Hmm, that all looks fine.
The only other time I have seen that error was in a workshop where people were loading phyloseq/taxmap objects from .RData files, but Im not sure that is the cause.
Can you send me the code you used to get to this point?
Unfortunately, I need to reproduce the bug before I can fix it.
I just tried it again in a brandnew Rproject with no other packages loaded and it worked. I guess one of the attached or loaded packages in my original session caused the problem. Here is the code with sessionInfo that worked for me. Maybe this helps in tracking down the bug.
Thanks for your help.
> p <- readRDS("path_to/pobj.rds")
Loading required package: phyloseq
> library(metacoder)
Loading required package: taxa
Attaching package: ‘taxa’
The following object is masked from ‘package:phyloseq’:
filter_taxa
This is metacoder verison 0.3.3 (stable)
> metaphyseq <- parse_phyloseq(p)
> metaphyseq
<Taxmap>
1284 taxa: aab. Bacteria ... bxk. TACGTAGGGGGCGA[truncated]
1284 edges: NA->aab, NA->aac, aab->aad, aab->aae, aab->aaf ... anw->bxg, anv->bxh, anu->bxi, axg->bxj, anu->bxk
4 data sets:
otu_table:
# A tibble: 680 x 52
taxon_id otu_id `1` `10` `11` `12` `13` `14` `15` `16` `17` `18` `19` `2` `20` `21` `22`
<chr> <chr> <int> <int> <int> <int> <int> <int> <int> <int> <int> <int> <int> <int> <int> <int> <int>
1 axh ASV1 23 19 28 40 20 77 94 45 22 21 35 30 37 99 47
2 axi ASV2 5 4 1 2 0 17 2 2 0 0 0 23 0 0 0
3 axj ASV3 5 6 0 19 5 6 18 1 0 7 4 1 3 4 0
# … with 677 more rows, and 35 more variables: `23` <int>, `24` <int>, `25` <int>, `26` <int>, `27` <int>,
# `28` <int>, `29` <int>, `3` <int>, `30` <int>, `31` <int>, …
tax_data:
# A tibble: 680 x 10
taxon_id otu_id Kingdom Phylum Class Order Family Genus Species ASV
<chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr>
1 axh ASV1 Bacteria Firmicu… Bacil… Bacil… Bacill… Virgiba… Genus_Virg… TACGTAGGGGGCAAGCGTTGTCCGGAATTAT…
2 axi ASV2 Bacteria Firmicu… Bacil… Bacil… Bacill… Virgiba… Genus_Virg… TACGTAGGGGGCGAGCGTTGTCCGGAATTAT…
3 axj ASV3 Bacteria Firmicu… Bacil… Bacil… Bacill… Ornithi… Genus_Orni… TACGTAGGGGGCAAGCGTTGTCCGGAATTAT…
# … with 677 more rows
sample_data:
# A tibble: 50 x 28
sample_id Sample.ID Treatment Days Strain Combo Moisture Salmonella pH Aw TOC TC IC TN
<chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr>
1 1 1 PLE 0 SH.28… PLE.… 0.182 6.055 8.06 0.824 2983… 3091… 1076… 7424…
2 10 10 PLE 14 SH.28… PLE.… 0.142 2.455 7.91 0.64 2082… 2168… 8623… 5547…
3 11 11 PLE 14 SH.28… PLE.… 0.165 2.428 7.91 0.646 2511… 2601… 9021… 5820…
# … with 47 more rows, and 14 more variables: NH4 <chr>, NO3 <chr>, Al <chr>, Ca <chr>, Cd <chr>, Cu <chr>,
# Fe <chr>, Mg <chr>, Mn <chr>, P <chr>, …
phy_tree:
Phylogenetic tree with 680 tips and 678 internal nodes.
Tip labels:
ASV1, ASV2, ASV3, ASV4, ASV5, ASV6, ...
Node labels:
, 0.872, 0.889, 0.000, 0.737, 0.857, ...
Unrooted; includes branch lengths.
0 functions:
> sessionInfo()
R version 3.6.1 (2019-07-05)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 18.04.3 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.7.1
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.7.1
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=de_AT.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=de_AT.UTF-8 LC_MESSAGES=en_US.UTF-8 LC_PAPER=de_AT.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C LC_MEASUREMENT=de_AT.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] metacoder_0.3.3 taxa_0.3.2 phyloseq_1.28.0
loaded via a namespace (and not attached):
[1] tidyselect_0.2.5 reshape2_1.4.3 purrr_0.3.2 splines_3.6.1 lattice_0.20-38 rhdf5_2.28.1
[7] colorspace_1.4-1 vctrs_0.2.0 stats4_3.6.1 mgcv_1.8-29 utf8_1.1.4 survival_2.44-1.1
[13] rlang_0.4.0 pillar_1.4.2 glue_1.3.1 BiocGenerics_0.30.0 foreach_1.4.7 plyr_1.8.4
[19] stringr_1.4.0 zlibbioc_1.30.0 Biostrings_2.52.0 munsell_0.5.0 gtable_0.3.0 codetools_0.2-16
[25] Biobase_2.44.0 permute_0.9-5 IRanges_2.18.3 biomformat_1.12.0 parallel_3.6.1 fansi_0.4.0
[31] Rcpp_1.0.2 scales_1.0.0 backports_1.1.5 vegan_2.5-6 S4Vectors_0.22.1 jsonlite_1.6
[37] XVector_0.24.0 ggplot2_3.2.1 stringi_1.4.3 dplyr_0.8.3 grid_3.6.1 ade4_1.7-13
[43] cli_1.1.0 tools_3.6.1 magrittr_1.5 lazyeval_0.2.2 tibble_2.1.3 cluster_2.1.0
[49] crayon_1.3.4 ape_5.3 pkgconfig_2.0.3 zeallot_0.1.0 MASS_7.3-51.4 Matrix_1.2-17
[55] data.table_1.12.4 assertthat_0.2.1 rstudioapi_0.10 iterators_1.0.12 Rhdf5lib_1.6.2 R6_2.4.0
[61] multtest_2.40.0 igraph_1.2.4.1 nlme_3.1-141 compiler_3.6.1
Hi! I noticed that in the example above you have 1284 taxa, but only 680 ASVs. Any idea of why there are more taxa than ASVs? I am running into the same problem with my dataset. Thanks!
hello @anaeiou,
The number of taxa and ASVs can be different. All ASVs could be assigned to a single taxon for example, or a single ASV could have a deep classification. For example, an ASV with the classification:
"Root;Actinobacteria;Actinobacteria;Actinomycetales;Nocardiaceae;Rhodococcus"
would be assigned to 6 taxa.
1284 taxa and 680 ASVs does not indicate a problem. I hope this clears things up some!
Thanks for the clarification! That makes sense- I was confused because in your tutorial (awesome resource, thank you so much for it!) you have 1,000 OTUs and they translate into 174 taxa even though each one has deep classification. Do you know why this might be? Either way, good to know I am (probably) not doing anything wrong :)
No problem! It depends on the data set and the taxonomy used. In the case of the example data:
library(metacoder)
x = parse_tax_data(hmp_otus, class_cols = "lineage", class_sep = ";",
class_key = c(tax_rank = "taxon_rank", tax_name = "taxon_name"),
class_regex = "^(.+)__(.+)$")
heat_tree(x, node_label = paste0(taxon_names, ' (', n_obs, ')'), node_size = n_obs, node_color = n_obs)

Created on 2021-04-12 by the reprex package (v0.3.0)
Note that many of the genera have lots of OTUs assigned to them (e.g., Bacterodies with 115).
Going back to the issue of displaying NAs:
I've been having this problem too. Unfortunately, I'm not able to reproduce it using reprex. However, I've figured out that this is only a problem for me when displaying taxmap objects within an R notebook but not in the console. Maybe this will be helpful for future debugging.
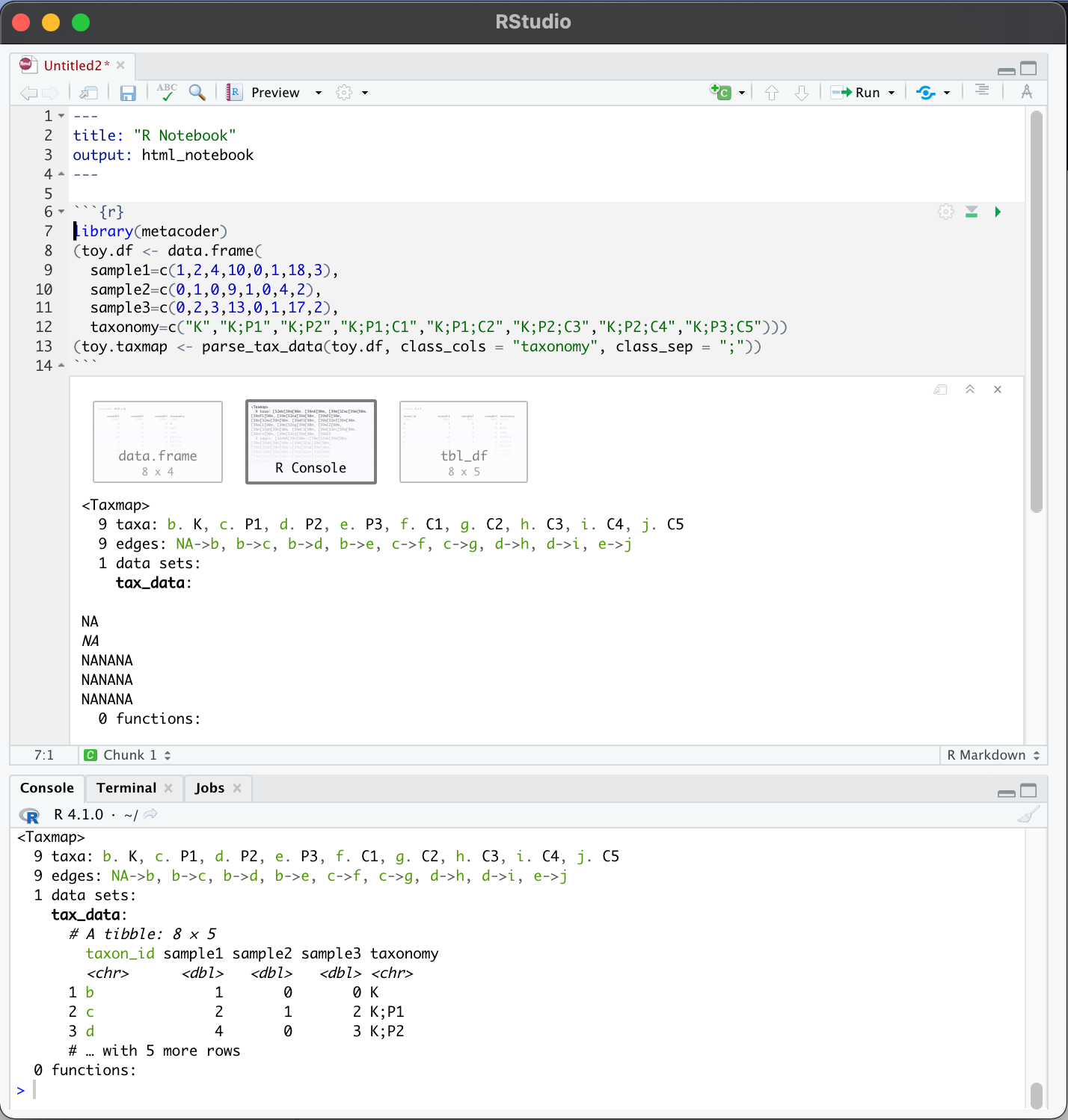
Yea, that looks like thats due to how Rmarkdown is rendered in Rstudio. Since that is an interaction with Rstudio, I am not sure how to fix it, but I will keep on the ook out for a solution. You can avoid that issue if you change this setting:
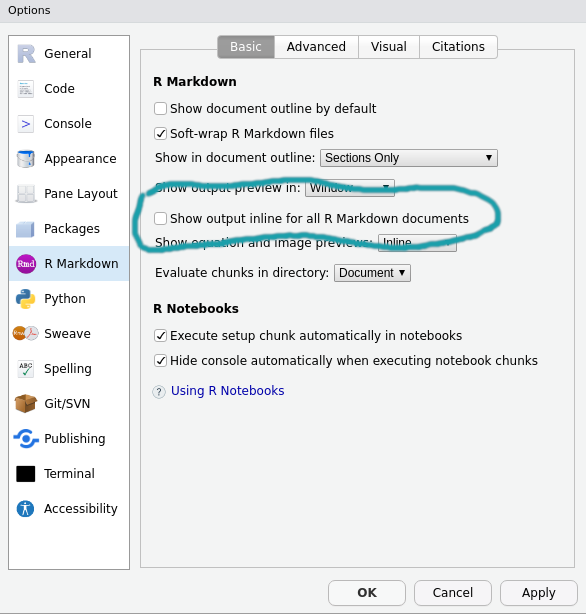
Which will cause the output to be printed to the console.