usegalaxy-playbook
 usegalaxy-playbook copied to clipboard
usegalaxy-playbook copied to clipboard
gtf sniffed as gff
Example files, uploaded via URL or browser:
- https://zenodo.org/record/290221/files/dexseq.gtf
- https://zenodo.org/record/290221/files/Drosophila_melanogaster.BDGP5.78.gtf
Test history:
- https://usegalaxy.org/u/jen/h/gtf-sniffed-as-gff
Tutorial:
- https://galaxyproject.github.io/training-material/topics/transcriptomics/tutorials/ref-based/tutorial.html
Data Source:
- https://zenodo.org/record/290221
Interestingly, gff3 is now accurately sniffed as gff3 (and not gff), also included in history for reference. This seems new but may not actually be new.
Tutorial:
- https://galaxyproject.github.io/training-material/topics/transcriptomics/tutorials/ref-based/tutorial.html
Data Source:
- https://zenodo.org/record/582600
ping @guerler
Is this Main-specific? It seems like an issue with the datatypes conf sniffer order or the sniffers themselves. Main has been using the sample datatypes_conf.xml since 5187d5de7e15e6cb5f375f34e9473c606e6973e8
Reran the tests, same result.
The problem is that the format of some datasets are a hybrid of GTF/GFF3 formatting - created by the DEXseq prepare annotation tool or manipulated datasets from other sources.
Line examples with mix of GFF/GFF3 and GTF style annotation in 9th group/attribute field:
Seqname Source Feature Start End Score Strand Frame Group 2L dexseq_prepare_annotation.py aggregate_gene 378112 387439 . + . gene_id "FBgn0000061" 2L dexseq_prepare_annotation.py exonic_part 378112 378481 . + . transcripts "FBtr0078053"; exonic_part_number "001"; gene_id "FBgn0000061" 2L dexseq_prepare_annotation.py exonic_part 384511 384894 . + . transcripts "FBtr0078053"; exonic_part_number "002"; gene_id "FBgn0000061" 2L dexseq_prepare_annotation.py exonic_part 385701 385746 . + . transcripts "FBtr0078053"; exonic_part_number "003"; gene_id "FBgn0000061" 2L dexseq_prepare_annotation.py exonic_part 386308 386576 . + . transcripts "FBtr0078053"; exonic_part_number "004"; gene_id "FBgn0000061" 2L dexseq_prepare_annotation.py exonic_part 386703 387439 . + . transcripts "FBtr0078053"; exonic_part_number "005"; gene_id "FBgn0000061"
Seqname Source Feature Start End Score Strand Frame Group chrUextra FlyBase gene 523024 523086 . - . gene_id "FBgn0264003"; gene_version "5"; gene_name "mir-5613"; gene_source "FlyBase"; gene_biotype "pre_miRNA"; chrUextra FlyBase transcript 523024 523086 . - . gene_id "FBgn0264003"; gene_version "5"; transcript_id "FBtr0330361"; transcript_version "5"; gene_name "mir-5613"; gene_source "FlyBase"; gene_biotype "pre_miRNA"; transcript_name "mir-5613-RM"; transcript_source "FlyBase"; transcript_biotype "pre_miRNA"; chrUextra FlyBase exon 523024 523086 . - . gene_id "FBgn0264003"; gene_version "5"; transcript_id "FBtr0330361"; transcript_version "5"; exon_number "1"; gene_name "mir-5613"; gene_source "FlyBase"; gene_biotype "pre_miRNA"; transcript_name "mir-5613-RM"; transcript_source "FlyBase"; transcript_biotype "pre_miRNA"; exon_id "FBtr0330361:1"; exon_version "1"; chrUextra FlyBase transcript 523024 523048 . - . gene_id "FBgn0264003"; gene_version "5"; transcript_id "FBtr0330363"; transcript_version "5"; gene_name "mir-5613"; gene_source "FlyBase"; gene_biotype "pre_miRNA"; transcript_name "mir-5613-RB"; transcript_source "FlyBase"; transcript_biotype "miRNA"; chrUextra FlyBase exon 523024 523048 . - . gene_id "FBgn0264003"; gene_version "5"; transcript_id "FBtr0330363"; transcript_version "5"; exon_number "1"; gene_name "mir-5613"; gene_source "FlyBase"; gene_biotype "pre_miRNA"; transcript_name "mir-5613-RB"; transcript_source "FlyBase"; transcript_biotype "miRNA"; exon_id "FBtr0330363:1"; exon_version "1";
There is not much that can be done - since it doesn't fit either GTF or GFF3 format (for a few reasons), the sniffer is falling back to the GFF format, which is the original with a looser specification - can put almost anything in the 9th "group/attribute" column and have what is technically a valid-ish GFF file for some use-cases.
Let's close this ticket out. If these datasets are only used with the intended tools, the assigned format is Ok. And is probably one reason why tools that consume these inputs are vague or combine "GFF3/GFF/GTF" format in the help text areas and associated tutorials. Some wrapped 3rd party tools produce custom hybrid datasets that are tool-group specific but close enough to other known datatypes that they are sniffed/assigned to them. This is how bioinformatics works in real life! :)
The only change that could be made is to rename the dataset at the source (especially if associated with our tutorials or publications) to have the .gff extension instead of gtf so that no one getting the data out of context thinks it is in GTF format. I'll open a ticket for this in training repo.
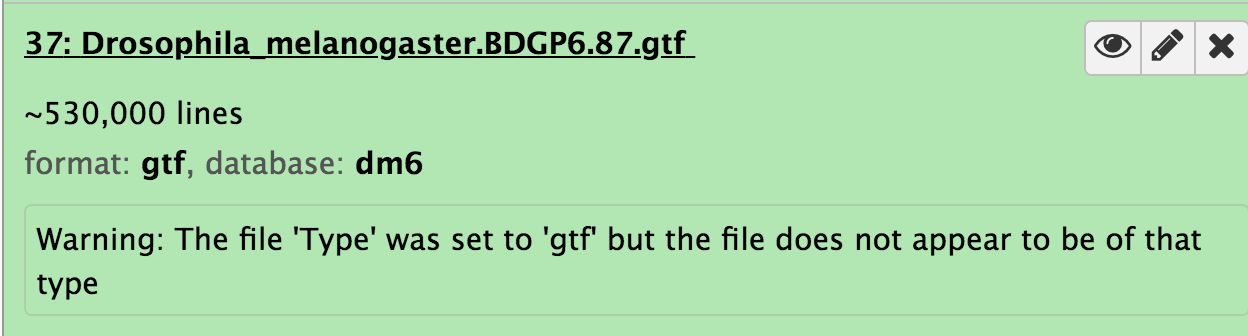
Ran into this issue (a GTF file sniffed as GFF) while testing the current ref-based RNA-seq tutorial using the GTF file provided in Zenodo (here: https://zenodo.org/record/1185122/files/Drosophila_melanogaster.BDGP6.87.gtf) in usegalaxy.eu.
And if I try with specifying the Type as "GTF" during upload I get a warning "Warning: The file 'Type' was set to 'gtf' but the file does not appear to be of that type" in the dataset in the history, see below.
Not sure where the Zenodo GTF file comes from, think Flybase or Ensembl.
@jennaj could you reopen this issue when you get a chance please as I'm seeing it with the current Ensembl GTF files e.g. Fly: ftp://ftp.ensembl.org/pub/release-92/gtf/drosophila_melanogaster/Drosophila_melanogaster.BDGP6.92.gtf.gz Human: ftp://ftp.ensembl.org/pub/release-92/gtf/homo_sapiens/Homo_sapiens.GRCh38.92.gtf.gz
The Ensembl GTFs are auto-importing as GFF, and if GTF is set as the Type on import, the warning appears that the file doesn't appear to be GTF. GTF is equivalent to GFF2 format but GTF format should take precedence here (and there shouldn't be a warning output saying this doesn't appear GTF format, it is GTF) imo. Is it a matter of reordering the GTF/GFF datatypes priority somewhere?
The first line of that file - parsed as:
[u'chr3R', u'FlyBase', u'gene', u'722370', u'722621', u'.', u'-', u'.', u'gene_id "FBgn0085804"; gene_name "CR41571"; gene_source "FlyBase"; gene_biotype "pseudogene";']
does not have a transcript_id so Galaxy does not sniff it as a gtf - https://github.com/galaxyproject/galaxy/blob/dev/lib/galaxy/datatypes/interval.py#L1059.
http://genome.ucsc.edu/FAQ/FAQformat#format4
The attribute list must begin with the two mandatory attributes:
gene_id value - A globally unique identifier for the genomic source of the sequence. transcript_id value - A globally unique identifier for the predicted transcript.
Rats. @mblue9 what do you think?
All these custom formats for GFF/GFF2/GTF/GFF3 are ... problematic
These data look like GFF3 that was modified to be in GTF format, but only some lines actually meet with the spec for GTF. Data also includes headers that do not fit GTF (should be none) or GFF3 (which wouldn't fit the content).
Loaded both into the history here: https://usegalaxy.org:/u/jen/h/test-gtf-downloads
What tools actually require can vary. Content and format both factors. We need to figure out a way to handle these somehow. Avoid the wrong datatype for format reasons (unexpected headers) and base it just on content. Then tools need to know how to handle that input based on content/datatype (ignore headers or we strip them out ourselves at Upload).
Or we just keep on defaulting back to a generic GFF and have users clean up the data. Which users have problems with. I don't know the right path for this data source. Some tools will work with it fine, some will not.