eggnog-mapper
 eggnog-mapper copied to clipboard
eggnog-mapper copied to clipboard
Fast genome-wide functional annotation through orthology assignment

when i run the command " emapper.py -i hap1_protein.fasfa --output xx1 --override --cpu 10 --decorate_gff xx.gff3" the xx.gff3 was maked by the evm and update with the funatation. but its...
its show the error with the bug 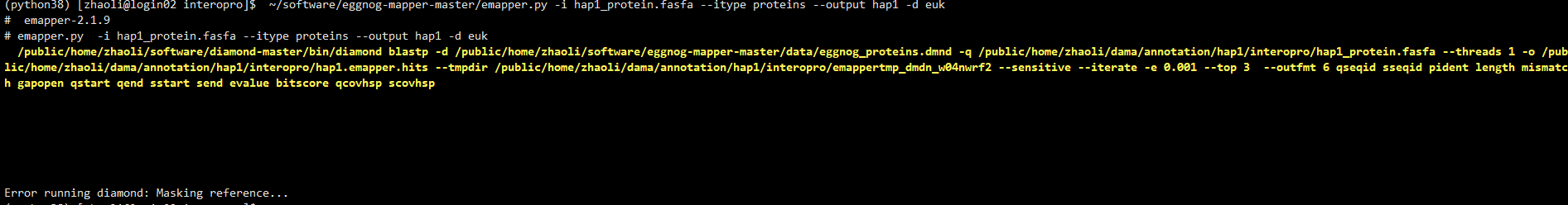 Error running diamond: Masking reference... how could I solve the error, thank you
Hi, Thanks for the great tool! For legacy reasons, is it possible to run emapper on eggnog_3.0? If yes, how to do so? Many thanks for your help!
Hi, I am trying to download all hmmer databases. Taxid 1 has the corrupted data, which could not be finished. All the rest taxid were well proceeded. Could you please...
Hi. I got puzzled when using do GO functional annotation to do enrichment. In many cases, one gene got a lot of GO ids in the results, more than the...
Hello, some tools uses `multiprocessing.cpu_count()` to ge the number of cpus. `multiprocessing.cpu_count()` return the number of cpu in the machine, But this is not the same as the number of...
Dear all, I want to build a personal species packages, and use for annotation with clusterProfiler. Since I run emapper.py with and without "--tax_scope" option, I got quite different results,...
Hello! I want to use eggnog-mapper (web-version) output to build genome-scale metabolic model via CarveMe tool so I am interested in BiGG reaction ids. For some reason no bigg reaction...
Hello! I'm trying to use eggnog mapper wep (http://eggnog-mapper.embl.de/). But there are two problems. 1. I was unable to submit genomic data successfully. (There is a circle turning all the...
Three error messages in emapper.py referenced "download_eggnog_database.py". It should be "download_eggnog_data.py"