`bamCompare --skipZeroOverZero` bugged
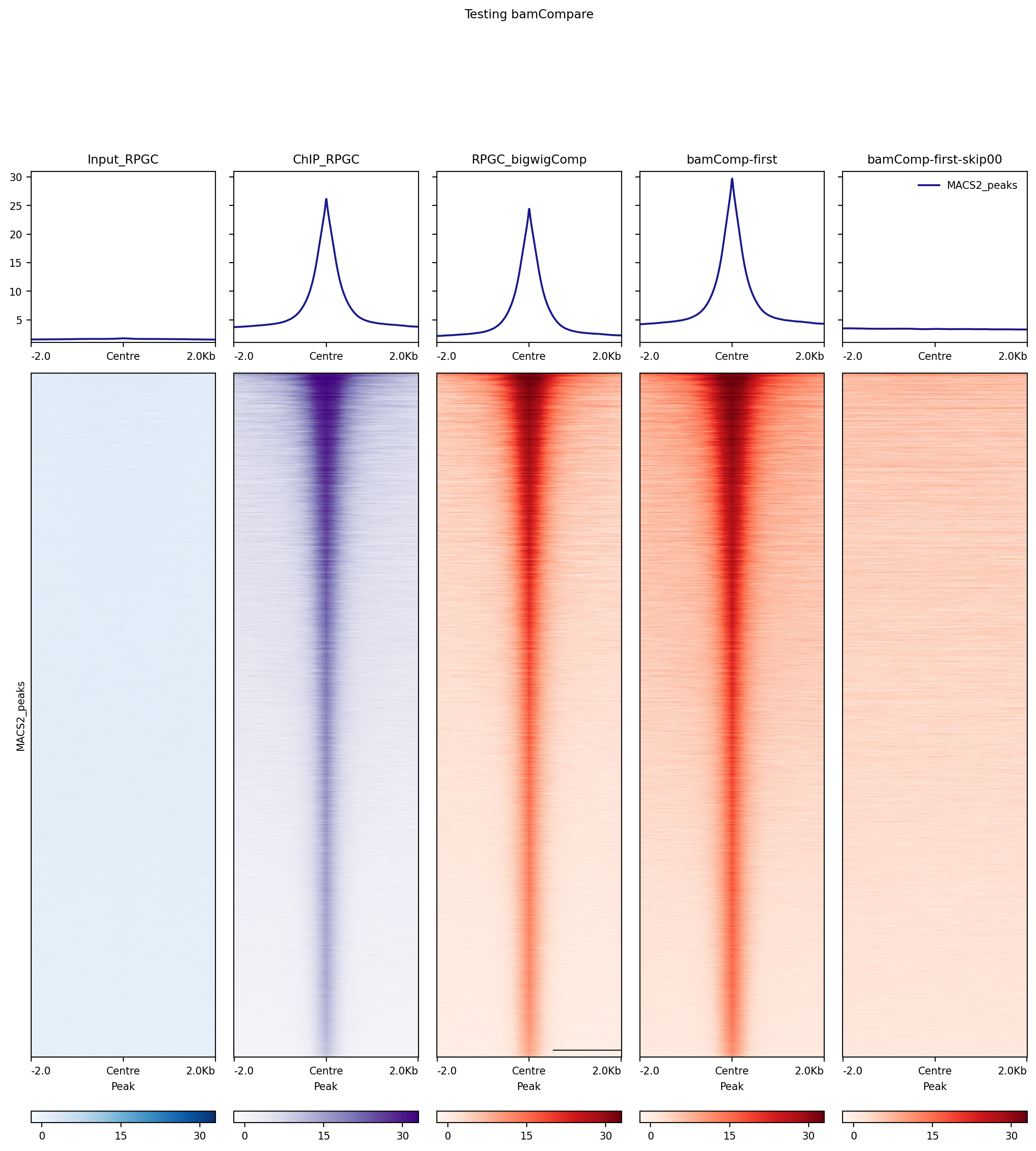
This is related to issue #1108. Running bamCompare with --skipZeroOverZero gives incorrect results. From left to right, the attached figure shows the following deepTools heatmaps: a ChIP-seq input control (RPGC-normalized output from bamCoverage); a ChIP-seq sample (RPGC-normalized output from bamCoverage); the output of bigwigCompare to subtract the input from the ChIP sample; the output of
bamCompare -b1 <sample> -b2 <input> --ignoreDuplicates --extendReads 200 --binSize 10 --scaleFactorsMethod None --normalizeUsing None --operation first -p 32;
and finally the output of the exact same bamCompare command as above with the addition of the --skipZeroOverZero flag. The peaks are clearly lost from the expected locations after skipping "0-over-0" bins. This could be explained by the coordinates being shifted after --skipZeroOverZero.
On lines 246-247 of writeBedGraph.py, I think value needs to be set to None (or possibly NaN) before continuing to the next iteration of the for loop. This will prevent situations where previous_value gets extended over non-covered regions. Not sure that this will fix the coordinate issue though, so perhaps there are two bugs here.
Having the same issue here. Using --skipZeroOverZero produces large bins that appear as long blocks in the UCSC browser when visualising the data. Removing --skipZeroOverZero resolves the issue.
Command used (deeptools v3.5.5):
bamCompare --bamfile1 sample1.bam \
--bamfile2 sample2.bam \
-o results.bg \
--outFileFormat bedgraph \
--binSize 100 \
--normalizeUsing CPM --scaleFactorsMethod None \
--effectiveGenomeSize $effectivegenomesize \
--ignoreForNormalization chrX chrY \
--extendReads \
--operation ratio \
--pseudocount 0 1 \
--skipZeroOverZero
Output bedgraph: