deepTools
 deepTools copied to clipboard
deepTools copied to clipboard
Published
20 hours ago •
 deeptools
deeptools
How to deal spike-in data correctly?
Hello!
I have a series of Chip-seq results.and I add some spike-in cells in my sample to normalized my chip valuel. I calculate the scale-factor from sample mapped reads and sipke-in mapped reads.and use Bamcoverage -scale to get bw files. But the meta plot shows that the corrected samples are significantly elevated overall, but the actual situation is that the reads near the genes in the samples are approximately the same. Is there any solution to this problem?
``
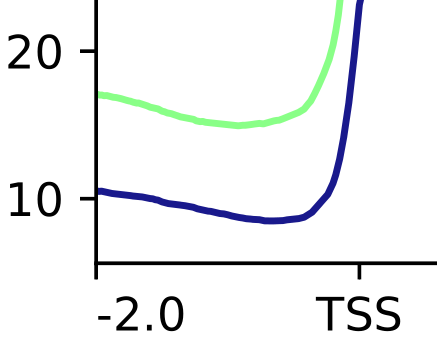 
Maybe you need to invert the scaling factors? What about signal in intergenic regions?