transformGamPoi-Paper
 transformGamPoi-Paper copied to clipboard
transformGamPoi-Paper copied to clipboard
[Question] How to benchmark another transformation
Hi @const-ae,
First up, love the paper!
I was wondering what is the best way to run your benchmarking on a single other transformation? I've implemented an empirical bayesian approach and want to see how it does if possible (https://github.com/mjcasy/scEC/blob/master/R/Normalisation.R).
Thanks
Hi mjcasy,
I was wondering what is the best way to run your benchmarking on a single other transformation?
The general steps are to
- Clone the
benchmarkfolder. - Implement a function that calls your
scECpackage in transformation_helper.R and add it to theall_transformationslist at the end of the scriptscEC_fnc <- function(UMI, sf, alpha){ scEC:::GetFreq(UMI) } all_transformations <- list(<...>, newwave = newwave_fnc, scEC = scEC_fnc) - Install the R dependencies by running
Rscript -e "renv::restore()"
The next step depends if you have access to a cluster that runs slurm.
If yes:
- Modify job_overview.yaml tos very slow transformation approaches.
- Set
.OUTPUT_FOLDERinrun_benchmarks.Rto an appropriate location - Run
Rscript src/run_benchmarks.Rto generate the jobs - Run
Rscript src/collect_results.Rto collect the results after all jobs have successfully finished
Alternatively, you can call the individual scripts to generate/download data, transform the data and evaluate the results manually. Let's say you want to check the consistency of your method:
- Prepare a results folder: e.g.
mkdir -p /tmp/transformation_benchmark/{results,duration,data} - Modify the
data_folderinconsistency_benchmark/download_helper. E.g.,data_folder <- "/tmp/transformation_benchmark/data" - Download the dataset
Rscript src/consistency_benchmark/download_10X_datasets.R \ --dataset GSE142647 \ --working_dir /tmp/transformation_benchmark \ --result_id GSE142647.Rds - Calculate both the k-NN graph for both gene halves
Rscript src/consistency_benchmark/transform_consistency_data.R \ --transformation scEC \ --input_id GSE142647.Rds \ --knn 50 \ --pca_dim 20 \ --alpha TRUE \ --seed 1 \ --working_dir /tmp/transformation_benchmark \ --result_id GSE142647_knns.Rds - Calculate the overlap of the two k-NN graphs
Rscript src/consistency_benchmark/calculate_10X_consistency.R \ --transformation scEC \ --input_id GSE142647_knns.Rds \ --dataset GSE142647 \ --knn 50 \ --pca_dim 20 \ --alpha TRUE \ --seed 1 \ --working_dir /tmp/transformation_benchmark \ --result_id GSE142647_consistency.tsv
You can now easily take these steps and wrap them in a for loop over seeds or datasets.
Let me know if you run into any trouble going through the steps. It is always an exciting moment when someone else is running one's code for the first time.
Thanks for the detailed response! Unfortunately the number of jobs launched hit up against admin limits on my institutions server, so will have to run without slurm. Any advice on what bits to run to get a slimmer result?
Oh, I ran into the same issue with the job limit. I submitted the jobs in batches of a few thousand. In addition, I usually changed the job priority to 'low' in the submission steps of run_benchmarks.R, as our clustered allowed several times more low priority jobs to be submitted.
If you want to reduce the number jobs, modify the job_overview.yaml. By default, each parameter combination mentioned in 'input_data' and 'knn_construction' is considered. So you can for example cut the number of consistency jobs by a factor of 10 if you remove half the datasets and only consider one seed (i.e., replicate). Or you focus only on the parameter combinations discussed in the manuscript, which is also only a fraction of the total 58,000 data points.
Send from my phone, so please excuse any typos.
Hi, thanks for your great work! I wonder if it is possible for me to only run certain methods (for example, GLM based model) on certain datasets. Thanks a lot.
Moreover, I also have a quick question about the benchmarking result:
Did you consider about problems related to cell type disappearing? For example, this dataset which is shown in GLM-PCA. It seems that changing the number of neighbors as a hyper parameter cannot resolve this problem.
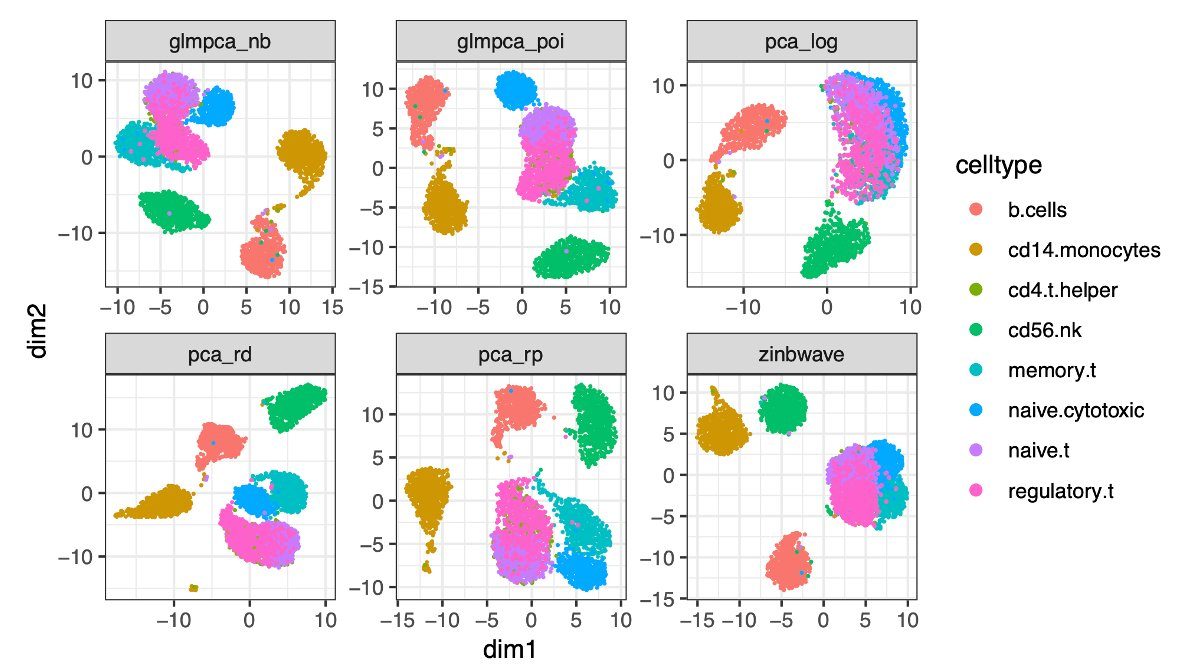
I wonder if it is possible for me to only run certain methods (for example, GLM based model) on certain datasets. Thanks a lot.
Yes! Take a look at the job_overview.yaml file. It declares which combination of dataset and transformation are benchmarked. You could for example remove all non-GLM-based models. The actual parsing of the file happens in run_benchmarks.R.
Did you consider about problems related to cell type disappearing? For example, this dataset which is shown in GLM-PCA. It seems that changing the number of neighbors as a hyper parameter cannot resolve this problem.
I am not 100% sure I understand the question. I did not specifically consider if cell types disappear (or maybe rather are mixed with other cell types) in the tSNE or UMAP plot. However, the overlapping kNN metric should capture the case if one method is able to better resolve the structure of a dataset (which has to be consistent with the ground truth structure).
Hi, yes, I initially agree with you about using KNN metric to evaluate it. However, do you think, given the plots I have in the paper GLM-PCA, the normalization method based on library size can reduce the resolution of cell types in the existing datasets? It is like a special case or boardline case, I wonder if there are any methods I can use to identify this case. Thanks.
Hi, another question, now I meet some errors to run Rscript -e "renv::restore()" in a new environment. May I know your R version? Thanks a lot.
Hi, actually plenty of errors. In most of the cases, its package installing error. I meet this error for stirngi, deseq2, biostringi, etc. Now I use R4.2.3 and I am keeping on trying.
Hi mjcasy,
I was wondering what is the best way to run your benchmarking on a single other transformation?
The general steps are to
- Clone the
benchmarkfolder.- Implement a function that calls your
scECpackage in transformation_helper.R and add it to theall_transformationslist at the end of the scriptscEC_fnc <- function(UMI, sf, alpha){ scEC:::GetFreq(UMI) } all_transformations <- list(<...>, newwave = newwave_fnc, scEC = scEC_fnc)- Install the R dependencies by running
Rscript -e "renv::restore()"The next step depends if you have access to a cluster that runs
slurm.If yes:
- Modify job_overview.yaml tos very slow transformation approaches.
- Set
.OUTPUT_FOLDERinrun_benchmarks.Rto an appropriate location- Run
Rscript src/run_benchmarks.Rto generate the jobs- Run
Rscript src/collect_results.Rto collect the results after all jobs have successfully finishedAlternatively, you can call the individual scripts to generate/download data, transform the data and evaluate the results manually. Let's say you want to check the consistency of your method:
- Prepare a results folder: e.g.
mkdir -p /tmp/transformation_benchmark/{results,duration,data}- Modify the
data_folderinconsistency_benchmark/download_helper. E.g.,data_folder <- "/tmp/transformation_benchmark/data"- Download the dataset
Rscript src/consistency_benchmark/download_10X_datasets.R \ --dataset GSE142647 \ --working_dir /tmp/transformation_benchmark \ --result_id GSE142647.Rds- Calculate both the k-NN graph for both gene halves
Rscript src/consistency_benchmark/transform_consistency_data.R \ --transformation scEC \ --input_id GSE142647.Rds \ --knn 50 \ --pca_dim 20 \ --alpha TRUE \ --seed 1 \ --working_dir /tmp/transformation_benchmark \ --result_id GSE142647_knns.Rds- Calculate the overlap of the two k-NN graphs
Rscript src/consistency_benchmark/calculate_10X_consistency.R \ --transformation scEC \ --input_id GSE142647_knns.Rds \ --dataset GSE142647 \ --knn 50 \ --pca_dim 20 \ --alpha TRUE \ --seed 1 \ --working_dir /tmp/transformation_benchmark \ --result_id GSE142647_consistency.tsvYou can now easily take these steps and wrap them in a for loop over seeds or datasets.
Hi, I can run your codes and add my own methods via R 4.2.3. Moreover, I wonder if I have my own downloaded datasets, what should I do to use it as a benchmark dataset. Thanks a lot!