mango
 mango copied to clipboard
mango copied to clipboard
run_example.sh woes on Amazon EMR
Hi, I built mango on Amazon EMR (5.3.1) as follows:
git clone https://github.com/bigdatagenomics/mango.git
cd mango
sh ./scripts/move_to_spark_2.sh
sh ./scripts/move_to_scala_2.11.sh
mvn clean package –DskipTests
The run-example script fails:
./example-files/run-example.sh
<snip>
17/09/13 14:42:34 INFO SparkContext: Created broadcast 0 from newAPIHadoopFile at ADAMContext.scala:945
Command body threw exception:
org.bdgenomics.mango.models.UnsupportedFileException: File type not supported. Stack trace: java.io.FileNotFoundException: Couldn't find any files matching example-files/ALL.chr17.7500000-7515000.phase3_shapeit2_mvncall_integrated_v5a.20130502.genotypes.vcf. If you are trying to glob a directory of Parquet files, you need to glob inside the directory as well (e.g., "glob.me.*.adam/*", instead of "glob.me.*.adam".
at org.bdgenomics.adam.rdd.ADAMContext.getFiles(ADAMContext.scala:413)
at org.bdgenomics.adam.rdd.ADAMContext.getFsAndFiles(ADAMContext.scala:443)
…
I found that by moving the example reads and variants files (but not the reference file) to hdfs, this problem was averted, and mango server starts.
bin/mango-submit ./example-files/hg19.17.2bit \
-genes http://www.biodalliance.org/datasets/ensGene.bb \
-reads hdfs:///example-files/chr17.7500000-7515000.bam.adam \
-variants hdfs:///example-files/ALL.chr17.7500000-7515000.phase3_shapeit2_mvncall_integrated_v5a.20130502.genotypes.vcf \
-show_genotypes \
-discover \
-port 8193
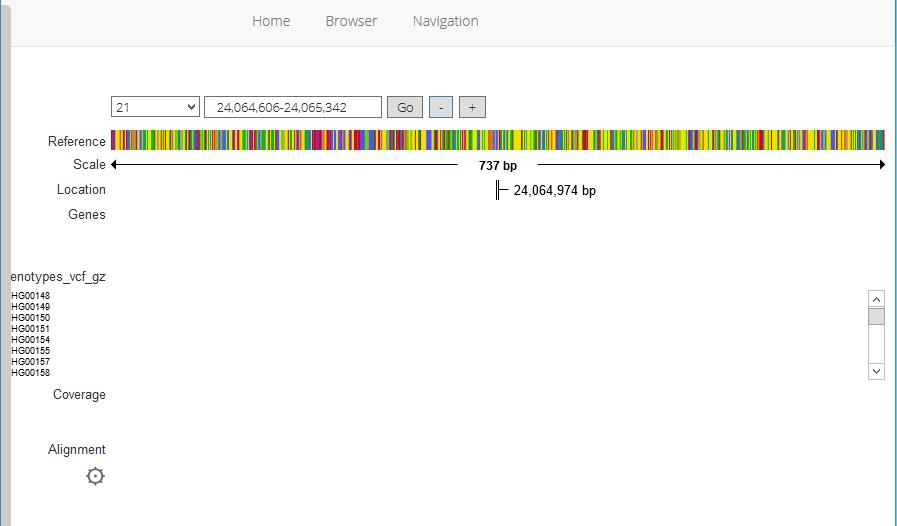
However, the browser doesn’t appear to be fully functional - see attached screenshot.
- It gets stuck on ‘loading Variants’
- It gets stuck on ‘Loading Genotypes’
- Coverage and Alignment sections are empty/non functional
- Variants file label truncated on the left
I’d be grateful for any pointers or workarounds.

Hi @rstrahan ! I would like to know if this is a data, data access or javascript issue. Could you:
- post any terminal output/errors
- look at the spark UI and see if jobs are being called
After these things, if we don't find anything, we can check the javascript for errors.
Hi @akmorrow13 - thanks for the speedy response! Logs attached:
-
run-example.log: terminal stdout/stderr associated with the first problem, where mango fails when using run-example.sh from the repo.
-
run-example-hdfs.log: terminal stdout/stderr associated with second problem - mango server and spark tasks are running, no errors noted, but the browser display has the problems I noted.
-
mango-browser-log.txt: firefox browser console showing mango errors
Hope this helps. Thanks for your help! Cheers Bob run-example.log.txt run-example-hdfs.log.txt mango-browser-log.txt
Ah yes, this seems to be an bug from updating ADAM version. I have made an issue and will address it in the next couple days https://github.com/bigdatagenomics/mango/issues/312
In terms of the truncated text, this has been addressed in pileup https://github.com/hammerlab/pileup.js/pull/458, so next time we update the module this will be fixed.
hi @rstrahan I believe I fixed these issues in https://github.com/bigdatagenomics/mango/pull/313 (except for the label truncation issue) Let me know if these updates fix your issue!
Thanks @akmorrow13. You latest commits resolved the problem when using the example-files, but only after copying them first to hdfs. I get the same error I originally reported if I try to run the original run-example.sh with the files on local disk.
Now that the example files work, I'm trying to get mango to work with 1000Genomes reference/BAM/VCF files.
- reference: I converted the 1000Genomes reference genome to ADAM format (in S3 bucket)
- reads: converted 1000Genomes BAM file to ADAM
- I noted your latest example files uses SAM rather than BAM for reads input - is this significant?
- variants: I tried using ADAM formatted variants, but mango reports errors reading expected VCF header
- does mango support ADAM formats for variants, or only VCF?
Here's what I'm trying:
bin/mango-submit \
s3://<mybucket>/reference/human_g1k_v37.adam/ \
-genes http://www.biodalliance.org/datasets/ensGene.bb \
-reads s3://<mybucket>/adam/bam=HG00154/ \
-variants s3://<mybucket>/vcfs/ALL.chr22.20130502.genotypes.vcf.gz \
-show_genotypes \
-port 8193 \
-discover
The BAM file has all contigs for HG00154, and the VCF has all samples for just chr22, but still, I expected to see variants genotype HG00154, and some coverage/alignment data, but these sections are blank.
This could be operator error on my part. Some questions:
- Can mango read all inputs directly from s3, or must files be staged first on HDFS?
- Can mango support reads input as ADAM file converted from BAM (using ADAM transformAlignments)?
- Can mango support variants input as ADAM file converted from VCF (with genotypes) (using ADAM transformVariants)?
Can you point me to any tutorials showing how to use adam & mango to prepare and browse large scale public genomics dataset like 1000 Genomes (s3://1000genomes)?
Thanks again for the fast fix, and looking forward to your pointers on the above. Cheers Bob
@rstrahan here are some (scattered) answers:
reads: converted 1000Genomes BAM file to ADAM I noted your latest example files uses SAM rather than BAM for reads input - is this significant?
No this is not, just easier for me to update changes with ADAM.
does mango support ADAM formats for variants, or only VCF? No but in the past there has been some issue with vcf.gz files. If you send me a reference to this vcf.gz file, I can test it out.
Can mango support reads input as ADAM file converted from BAM (using ADAM transformAlignments)? Yes, mango can read both ADAM and BAM.
Can mango support variants input as ADAM file converted from VCF (with genotypes) (using ADAM transformVariants)? Yes, mango supports both .vcf and .vcf.adam files for variants.
Can you point me to any tutorials showing how to use adam & mango to prepare and browse large scale public genomics dataset like 1000 Genomes (s3://1000genomes)?
I will put together a tutorial in the next couple weeks for this!