dada2
 dada2 copied to clipboard
dada2 copied to clipboard
RevComp primers remaining in some reads after trimming
I am trimming primers and truncating using the filterAndTrim function and am finding that I have some reverse compliment primer left in the reads. I am working with V4 region data using 515F-806R primers. The samples were sequenced using MiSeq 2 x 300 bp.
These are the trim parameters I'm using:
out <- filterAndTrim(list16S_Fs, fnFs.trim, list16S_Rs, fnRs.trim, trimLeft = c(19,20), truncLen=c(260,240), maxN=0, maxEE=c(2,2), truncQ=0, rm.phix=TRUE, compress=TRUE, multithread=3, matchIDs=TRUE)
I find that a small number (~3-10) of reverse reads in each sample contain the reverse compliment of the forward primer, and a slightly larger number (~60-80) of forward reads in each sample contain the reverse compliment of the reverse primer. I have also tried adding trimRight to also remove 20 bp from the right side both fwd and rev sequences, just to see if this removes these remaining primers, and it doesn't.
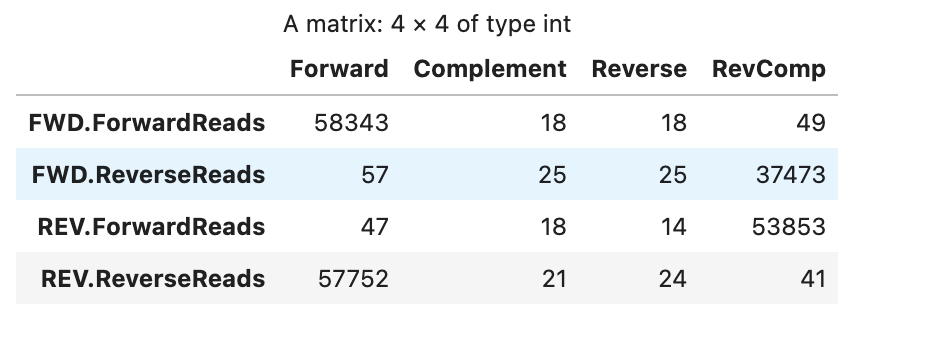
Before trimming
after trimming
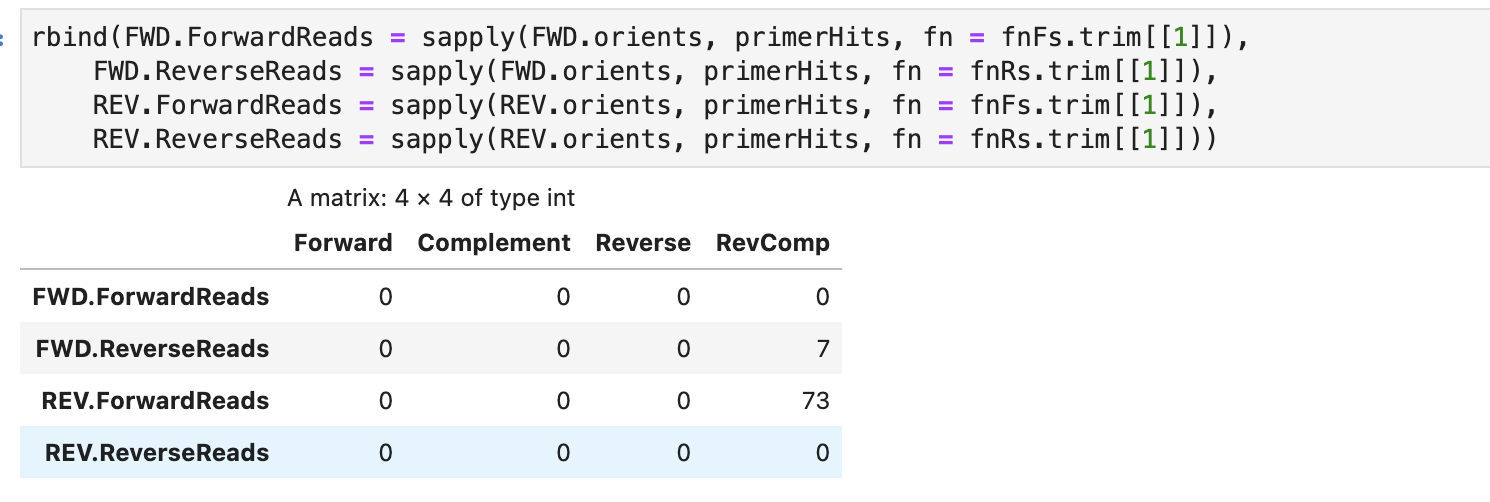
Is this normal? Can I remove this primers using filterAndTrim (and do I have to)? Should I try to trim much more aggressively? Even though the quality of the reads looks good and I otherwise wouldn't trim more, I have wiggle room to still be able to join the reads. I also have a similar issue with small numbers of reverse reads containing forward primer and forward reads containing reverse primer after primer removal via cutadapt from ITS region sequencing on the same samples from the same genomics center.
An example sequence is attached. The primer sequences are: fwd <- "GTGCCAGCMGCCGCGGTAA" rev <- "GGACTACHVGGGTWTCTAAT"
Thanks for your time! Emma 101PARCE2021_16S_S1_R1_001.fastq.gz 101PARCE2021_16S_S1_R2_001.fastq.gz
First, are the primers sequenced here? In the most common V4 16S libary setups they aren't. If they aren't you should not be removing any b ases from the start of the reads, that is trimLeft should be zero (the default).
Second, the length of this sequencing amplicon is (usually, in library preps that don't sequence the primers) 251-256 bps. So, you have to truncate your reads shorter than that or you are reading into the primers/adapters on the other side.
Finally, a tiny number of remaining opposite end primers is not a cause for concern. In this case it might have put up a flag on your not truncating your reads short enough, so silver linining, but in general that wouldn't be any cause for concern.
Hi, thanks for your support with this question and beyond! I have truncated my reads to the expected amplicon length accordingly.
In response to your question about whether the primers are sequenced here, I think that they are? The library was prepped using amplicon-indexing for sequencing on Illumina MiSeq, and my impression was that this method sequences primers. I also checked to see if there were primer hits in the raw reads I received, and there were (first table in my post). When I trimmed left by the number of bp in the primers, there were no longer primer hits. Therefore, I assumed that the primers were sequenced, and I proceeded with the trimmed reads. Please let me know if this logic is incorrect.
I also checked to see if there were primer hits in the raw reads I received, and there were (first table in my post). When I trimmed left by the number of bp in the primers, there were no longer primer hits.
Yep, that makes sense! I'm just used to seeing V4 data w/o primers, and glossed over the data you provided.