Questions about `mp_import_dada2` and `mp_cal_rarecurve` function
seqtab_no_chimeras_filtlen.rds.txt taxa_gtdb.rds.txt metadata_full_dada2_simple.txt
seqtabfile <- "seqtab_no_chimeras_filtlen.rds.txt"
taxafile <- "taxa_gtdb.rds.txt"
samplefile <- "metadata_full_dada2_simple.txt"
seqtab <- readRDS(seqtabfile)
taxa <- readRDS(taxafile)
mpse <- mp_import_dada2(seqtab=seqtab,
taxatab=taxa,
sampledata=samplefile)
mpse@colData
DataFrame with 72 rows and 0 columns
It is weird, On my platform, it contains the metadata.
> library(MicrobiotaProcess)
MicrobiotaProcess v1.9.0.990 For help:
https://github.com/YuLab-SMU/MicrobiotaProcess/issues
If you use MicrobiotaProcess in published research, please cite the
paper:
S Xu, L Zhan, W Tang, Z Dai, L Zhou, T Feng, M Chen, S Liu, X Fu, T Wu,
E Hu, G Yu. MicrobiotaProcess: A comprehensive R package for managing
and analyzing microbiome and other ecological data within the tidy
framework. 04 February 2022, PREPRINT (Version 1) available at Research
Square [https://doi.org/10.21203/rs.3.rs-1284357/v1]
This message can be suppressed by:
suppressPackageStartupMessages(library(MicrobiotaProcess))
> seqtabfile <- "seqtab_no_chimeras_filtlen.rds.txt"
> taxafile <- "taxa_gtdb.rds.txt"
> samplefile <- "metadata_full_dada2_simple.txt"
> seqtab <- readRDS(seqtabfile)
> taxa <- readRDS(taxafile)
> mpse <- mp_import_dada2(seqtab=seqtab, taxatab=taxa, sampleda=samplefile)
> mpse
# A MPSE-tibble (MPSE object) abstraction: 1,541,808 × 11
# OTU=21414 | Samples=72 | Assays=Abundance | Taxonomy=Kingdom, Phylum, Class, Order, Family, Genus, Species
OTU Sample Abundance dolphin_species Kingdom Phylum Class Order Family
<chr> <chr> <int> <chr> <chr> <chr> <chr> <chr> <chr>
1 OTU_1 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
2 OTU_2 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
3 OTU_3 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
4 OTU_4 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
5 OTU_5 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
6 OTU_6 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
7 OTU_7 HKWHF0621… 41 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
8 OTU_8 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
9 OTU_9 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
10 OTU_10 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
# … with 1,541,798 more rows, and 2 more variables: Genus <chr>, Species <chr>
> mpse %>% mp_extract_sample
# A tibble: 72 × 2
Sample dolphin_species
<chr> <chr>
1 HKWHF0621AM1_S1 Neophocaena phocaenoides
2 HKWHF0621AM10_S10 Neophocaena phocaenoides
3 HKWHF0621AM11_S11 Neophocaena phocaenoides
4 HKWHF0621AM12_S12 Neophocaena phocaenoides
5 HKWHF0621AM13_S13 Neophocaena phocaenoides
6 HKWHF0621AM14_S14 Neophocaena phocaenoides
7 HKWHF0621AM15_S15 Neophocaena phocaenoides
8 HKWHF0621AM16_S16 Neophocaena phocaenoides
9 HKWHF0621AM17_S17 Neophocaena phocaenoides
10 HKWHF0621AM18_S18 Neophocaena phocaenoides
# … with 62 more rows
> mpse@colData
DataFrame with 72 rows and 1 column
dolphin_species
<character>
HKWHF0621AM1_S1 Neophocaena phocaeno..
HKWHF0621AM10_S10 Neophocaena phocaeno..
HKWHF0621AM11_S11 Neophocaena phocaeno..
HKWHF0621AM12_S12 Neophocaena phocaeno..
HKWHF0621AM13_S13 Neophocaena phocaeno..
... ...
HKWHF0621AM70_S70 Neophocaena phocaeno..
HKWHF0621AM71_S71 Neophocaena phocaeno..
HKWHF0621AM72_S72 Neophocaena phocaeno..
HKWHF0621AM8_S8 Sousa chinensis
HKWHF0621AM9_S9 Neophocaena phocaeno..
> sampleda <- MicrobiotaProcess:::read_qiime_sample(samplefile)
> mpse2 <- mp_import_dada2(seqtab=seqtab, taxatab=taxa, sampleda=sampleda)
> mpse2
# A MPSE-tibble (MPSE object) abstraction: 1,541,808 × 11
# OTU=21414 | Samples=72 | Assays=Abundance | Taxonomy=Kingdom, Phylum, Class, Order, Family, Genus, Species
OTU Sample Abundance dolphin_species Kingdom Phylum Class Order Family
<chr> <chr> <int> <chr> <chr> <chr> <chr> <chr> <chr>
1 OTU_1 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
2 OTU_2 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
3 OTU_3 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
4 OTU_4 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
5 OTU_5 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
6 OTU_6 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
7 OTU_7 HKWHF0621… 41 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
8 OTU_8 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
9 OTU_9 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
10 OTU_10 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
# … with 1,541,798 more rows, and 2 more variables: Genus <chr>, Species <chr>
> mpse2 %>% mp_extract_sample
# A tibble: 72 × 2
Sample dolphin_species
<chr> <chr>
1 HKWHF0621AM1_S1 Neophocaena phocaenoides
2 HKWHF0621AM10_S10 Neophocaena phocaenoides
3 HKWHF0621AM11_S11 Neophocaena phocaenoides
4 HKWHF0621AM12_S12 Neophocaena phocaenoides
5 HKWHF0621AM13_S13 Neophocaena phocaenoides
6 HKWHF0621AM14_S14 Neophocaena phocaenoides
7 HKWHF0621AM15_S15 Neophocaena phocaenoides
8 HKWHF0621AM16_S16 Neophocaena phocaenoides
9 HKWHF0621AM17_S17 Neophocaena phocaenoides
10 HKWHF0621AM18_S18 Neophocaena phocaenoides
# … with 62 more rows
> mpse2@colData
DataFrame with 72 rows and 1 column
dolphin_species
<character>
HKWHF0621AM1_S1 Neophocaena phocaeno..
HKWHF0621AM10_S10 Neophocaena phocaeno..
HKWHF0621AM11_S11 Neophocaena phocaeno..
HKWHF0621AM12_S12 Neophocaena phocaeno..
HKWHF0621AM13_S13 Neophocaena phocaeno..
... ...
HKWHF0621AM70_S70 Neophocaena phocaeno..
HKWHF0621AM71_S71 Neophocaena phocaeno..
HKWHF0621AM72_S72 Neophocaena phocaeno..
HKWHF0621AM8_S8 Sousa chinensis
HKWHF0621AM9_S9 Neophocaena phocaeno..
>
or you can add the metadata using left_join
> mpse3 <- mp_import_dada2(seqtab=seqtab, taxatab=taxa)
> sampleda <- MicrobiotaProcess:::read_qiime_sample(samplefile)
> sampleda %<>% tibble::as_tibble(rownames='Sample')
> mpse3 %>% left_join(sampleda, by='Sample')
# A MPSE-tibble (MPSE object) abstraction: 1,541,808 × 11
# OTU=21414 | Samples=72 | Assays=Abundance | Taxonomy=Kingdom, Phylum, Class, Order, Family, Genus, Species
OTU Sample Abundance dolphin_species Kingdom Phylum Class Order Family
<chr> <chr> <int> <chr> <chr> <chr> <chr> <chr> <chr>
1 OTU_1 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
2 OTU_2 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
3 OTU_3 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
4 OTU_4 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
5 OTU_5 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
6 OTU_6 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
7 OTU_7 HKWHF0621… 41 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
8 OTU_8 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
9 OTU_9 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
10 OTU_10 HKWHF0621… 0 Neophocaena ph… k__Bac… p__Fi… c__C… o__P… f__Pe…
# … with 1,541,798 more rows, and 2 more variables: Genus <chr>, Species <chr>
Dear @xiangpin,
when I call mp_cal_rarecurve using above data, it generated error like below:
Error in seq.default(0, sdepth, by = step) : invalid '(to - from)/by'
Do you have a similar issue ?
I think this is because the total Abundance of some samples is too low compared with others. It seems some issues happened in the upstream analysis (ASV analysis or OTU clustering) if the samples are from a similar environment.
> seqtabfile <- "seqtab_no_chimeras_filtlen.rds.txt"
> taxafile <- "taxa_gtdb.rds.txt"
> samplefile <- "metadata_full_dada2_simple.txt"
> seqtab <- readRDS(seqtabfile)
> taxa <- readRDS(taxafile)
> mpse <- mp_import_dada2(seqtab=seqtab, taxatab=taxa, sampleda=samplefile)
> mpse %>% mp_extract_assays(.abundance=Abundance) %>% colSums() %>% sort()
HKWHF0621AM33_S33 HKWHF0621AM52_S52 HKWHF0621AM44_S44 HKWHF0621AM11_S11
64 304 23976 29861
HKWHF0621AM25_S25 HKWHF0621AM20_S20 HKWHF0621AM13_S13 HKWHF0621AM31_S31
31793 36487 36600 37964
HKWHF0621AM12_S12 HKWHF0621AM10_S10 HKWHF0621AM5_S5 HKWHF0621AM17_S17
38383 39363 41193 41677
HKWHF0621AM49_S49 HKWHF0621AM34_S34 HKWHF0621AM28_S28 HKWHF0621AM51_S51
44053 44137 44831 44979
HKWHF0621AM29_S29 HKWHF0621AM4_S4 HKWHF0621AM9_S9 HKWHF0621AM36_S36
48711 48908 49868 50469
HKWHF0621AM42_S42 HKWHF0621AM41_S41 HKWHF0621AM50_S50 HKWHF0621AM14_S14
50500 50679 53393 54078
HKWHF0621AM24_S24 HKWHF0621AM15_S15 HKWHF0621AM72_S72 HKWHF0621AM22_S22
54187 54825 54891 55004
HKWHF0621AM8_S8 HKWHF0621AM3_S3 HKWHF0621AM19_S19 HKWHF0621AM45_S45
55317 56064 56238 57540
HKWHF0621AM55_S55 HKWHF0621AM53_S53 HKWHF0621AM2_S2 HKWHF0621AM18_S18
59154 63628 63673 63840
HKWHF0621AM6_S6 HKWHF0621AM46_S46 HKWHF0621AM58_S58 HKWHF0621AM35_S35
65793 66671 68505 69832
HKWHF0621AM57_S57 HKWHF0621AM67_S67 HKWHF0621AM26_S26 HKWHF0621AM68_S68
70723 71560 71713 72159
HKWHF0621AM63_S63 HKWHF0621AM65_S65 HKWHF0621AM43_S43 HKWHF0621AM30_S30
72358 73592 74350 76080
HKWHF0621AM7_S7 HKWHF0621AM47_S47 HKWHF0621AM23_S23 HKWHF0621AM1_S1
76710 76871 76963 77822
HKWHF0621AM54_S54 HKWHF0621AM60_S60 HKWHF0621AM66_S66 HKWHF0621AM64_S64
79839 80186 80403 80969
HKWHF0621AM71_S71 HKWHF0621AM27_S27 HKWHF0621AM37_S37 HKWHF0621AM48_S48
84269 85475 86706 91891
HKWHF0621AM70_S70 HKWHF0621AM21_S21 HKWHF0621AM40_S40 HKWHF0621AM16_S16
94888 95254 96400 100201
HKWHF0621AM59_S59 HKWHF0621AM39_S39 HKWHF0621AM69_S69 HKWHF0621AM61_S61
104488 110443 112426 118551
HKWHF0621AM38_S38 HKWHF0621AM62_S62 HKWHF0621AM32_S32 HKWHF0621AM56_S56
128933 134000 175985 326983
I have fixed the issue, but I think this is better to check the samples or upstream pipeline.
mpse %<>% mp_cal_rarecurve(.abundance=Abundance, force=T)
mpse %>% mp_plot_rarecurve(.rare=AbundanceRarecurve, .alpha=Observe, .group=dolphin_species)
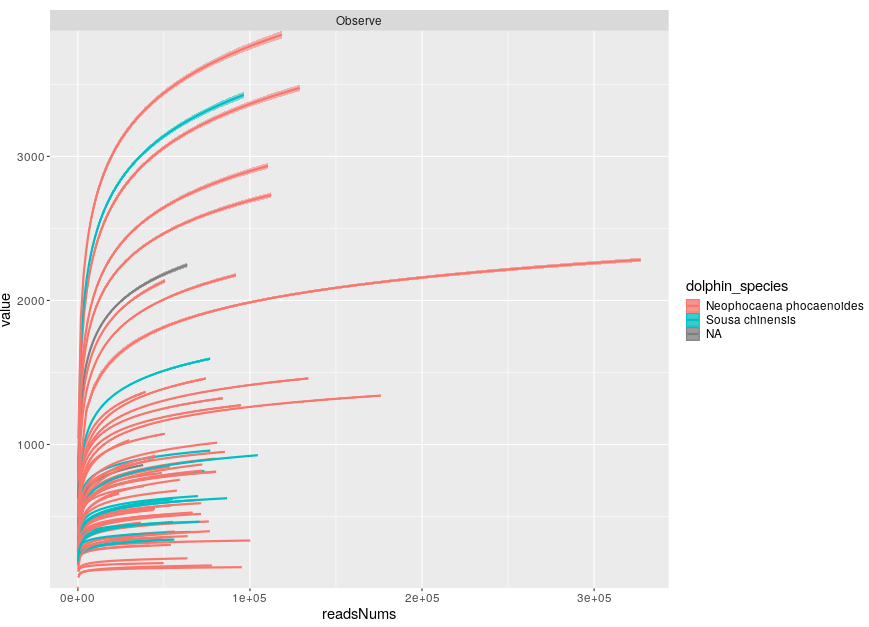
Great! Thanks for your analysis. It is true that some samples have too few ASVs.