GetOrganelle
 GetOrganelle copied to clipboard
GetOrganelle copied to clipboard
Incomplete chloroplast assembly/ a set of contigs instead of circular chloroplast sequence
Hi,
I used GetOrganelle to assemble chloroplast genomes and for two plant species, only one output is given and they do not appear as complete sequences, instead, seven and three contigs are given for each species. could you please let me know what parameters should I change to re-run the analysis? it is mentioned in this page that we can change -w and run the analysis, what is the range of values that we can apply for -w? For my analysis, w=74 has been selected by default since we did not set any value.
I have attached two output log files for your kind perusal. Please let me know what should I do to improve this chloroplast sequence .sylvatica_get_org.log.txt get_org.log.txt
You should first determine whether it is organelle-sufficient graph. If you cannot determine, please attach the fastg-based graph file (can be png visualized using Bandage for data privacy) here.
For determine organelle-sufficient graph, you may also refer to https://github.com/Kinggerm/GetOrganelle/issues/122#issuecomment-1005368502
hello,I have similar probloms with this issues, the initial assembly drawing is as follows, this is log
log.txt
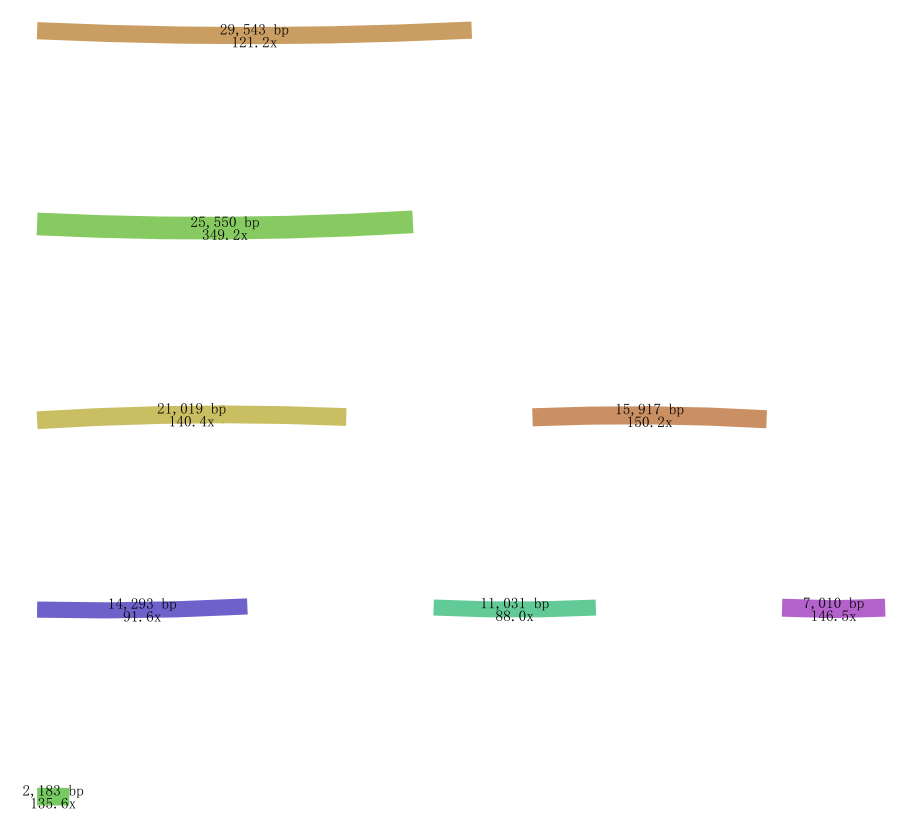
i rewrite -w 85 --max-reads inf --reduce-reads-for-coverage inf ,The result still is scaffolds,could you give me some advise?
@kibzhulab
Thanks for reaching out and for following the right proper thread.
Please visualize the *.fastg file rather than the *.gfa file. Please provide the visualization of *.fastg file for both the default parameter set result and the revised parameter set "-w 85 --max-reads inf --reduce-reads-for-coverage inf" result if possible.
thanks for your reply, this is my result from default parameter
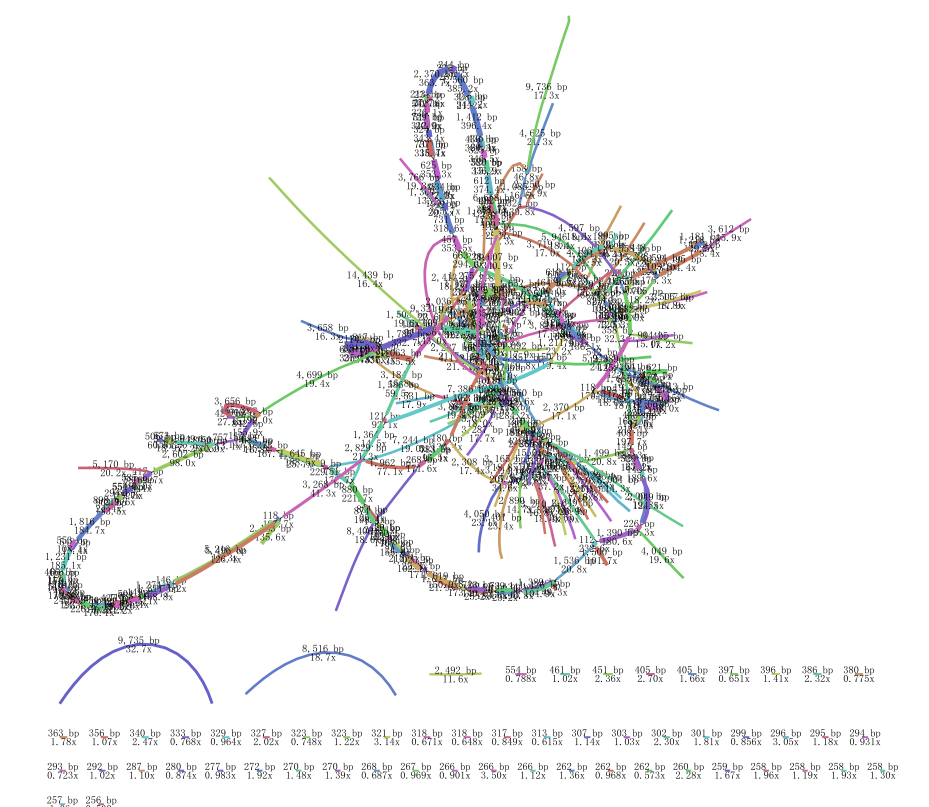 there is the log file
get_org.log.txt
there is the log file
get_org.log.txt
this is my another file from revised parameter set "-w 85 --max-reads inf --reduce-reads-for-coverage inf",the graf seems to be more complicated and mess.
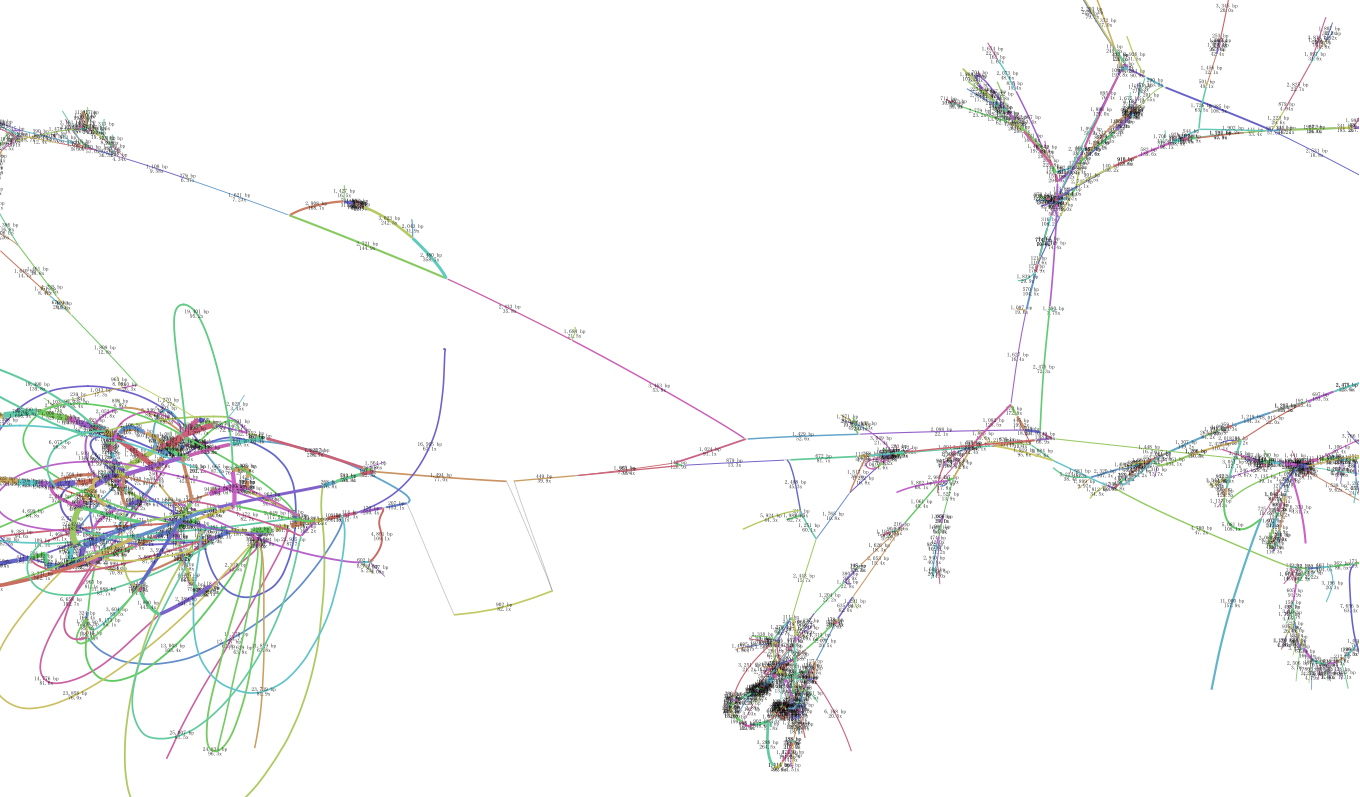 there is the log file
get_org.log.txt
there is the log file
get_org.log.txt
Those from 100x through 300x depth contigs are more likely to be the embplant_pt contigs. You can set depth range in the Bandage, e.g. 50x ~ 500x to see the remaining draftly filtered graph to determine whether they are organelle-sufficient. The default parameter one seems not because some target contigs are losing connections from one end. I cannot tell of the revised one because it's a little messy and not sharp.