smudgeplot
 smudgeplot copied to clipboard
smudgeplot copied to clipboard
long run time
Hi, my smudgeplot.py hetkmers job crashed multiple times because I ran out of memory. I manged to finally get it running by exclusively reserving an entire high memory node on our server (1.5Tb). It now appears to be running fine, but it is only using a single thread and has been running for 5 days. How long should I expect the job to run?
I am using ~75 Gb of CCS data. I couldn't get tbenavi1/KMC to compile due to dependency issues, so I used a different version of KMC.
Here are the commands I have run so far:
kmc -k21 -t20 -m1200 -ci1 -cs10000000 @FILES kmer_counts tmp/
kmc_tools transform kmer_counts histogram kmer_k21_full_with_zeros.hist -cx10000000
awk '{ if( $2 != 0 ){ print $0 } }' kmer_k21_full_with_zeros.hist > kmer_k21_full.hist && rm kmer_k21_full_with_zeros.hist
/project/ag100pest/software/genomescope2.0/genomescope.R -i kmer_k21_full.hist -k 21 -p 3 -l 9 -o genomescope_high_coverage_set_cov_prior_triploid -n tick
L=$(smudgeplot.py cutoff kmer_k21_full.hist L)
U=$(smudgeplot.py cutoff kmer_k21_full.hist U)
echo $L $U
kmc_tools transform kmer_counts -ci"$L" -cx"$U" dump -s kmer_counts_L"$L"_U"$U".dump
smudgeplot.py hetkmers -o kmer_counts_L"$L"_U"$U" < kmer_counts_L"$L"_U"$U".dump
L = 10 U = 690 My kmer_counts_L10_U690.dump file is 48 Gb.
As background, this is a tick species that has both diploid and triploid lineages and all indications are that we are sequencing from a triploid population. Although we have ~75 Gb of CCS data, the best GenomeScope plot I could generate had a triploid model with coverage prior of 9. Other models I generated that didn't specify a triploid model and a coverage prior didn't properly capture the first peak. None of the models fully captured the height of the first peak. A kcov of 9 is in agreement with the hom_peak of 9 found by HiFiASM. Given the estimated genome size however I would have expected to have ~30x coverage.
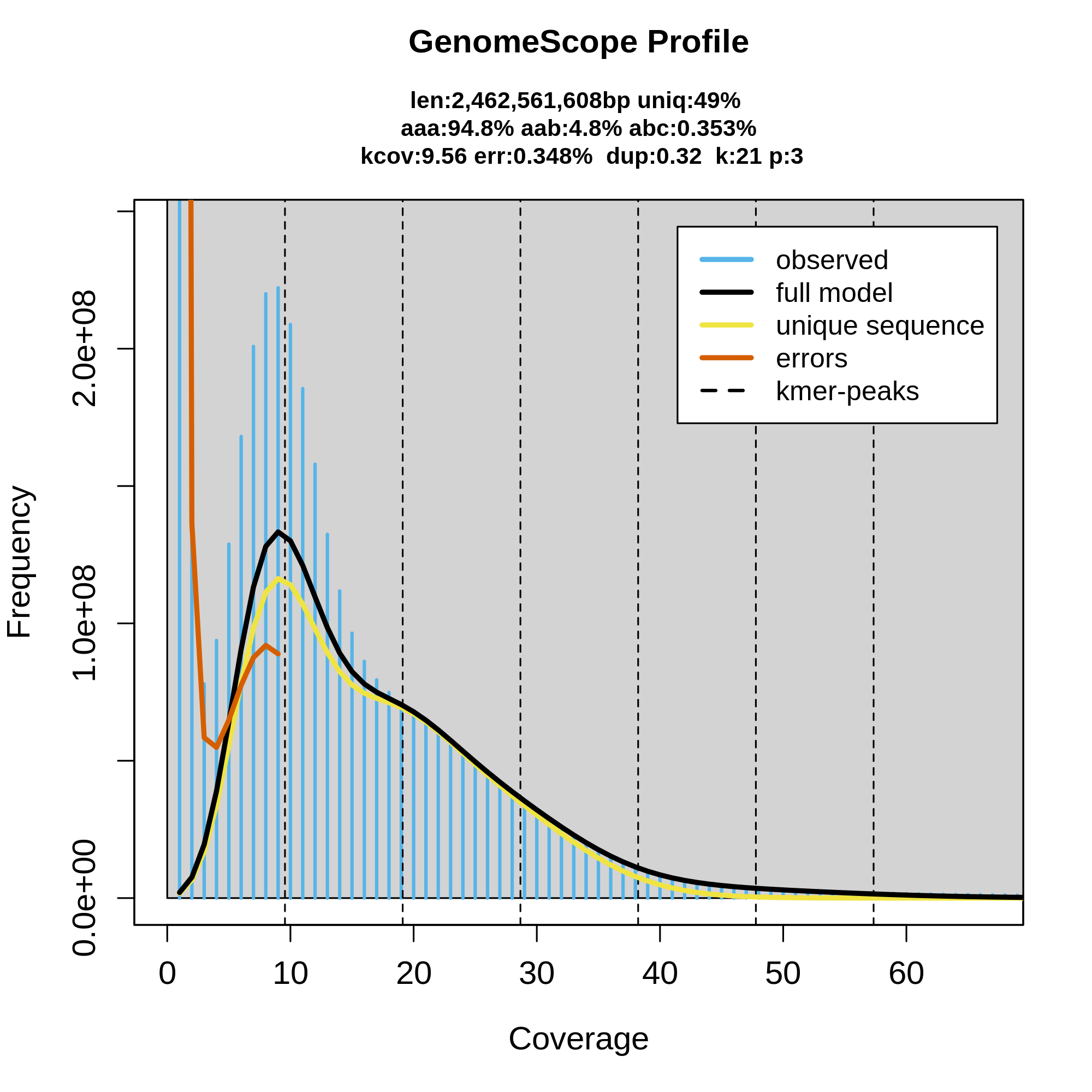
I'd appreciate any insights you might have based on the GenomeScope plot as well as any thoughts on how long I should expect the hetkmers job to run (or how I could speed it up). Thanks!
Hi Anna,
first of all - this looks like a really cool genome. Do I get right you though the genome to me a lot smaller than 2.5G? The confusing thing with this genome is that 1n peak is a lot higher than 2n peak (each position of AAB type should generate one of each), which would suggest that all the three haplotypes in the genome (assuming it is indeed triploid) are really divergent from each other (a few %), perhaps including some larger scale deletions/aneuploidy (which would make explanation of the 1n peak a bit easier). The only spectra I have seen like this are Meloidogyne root knot nematodes (and they do all the things mentioned above).
So, yeah, it would be great to see a smudgeplot of this one!
How long should I expect the job to run?
This depends how many k-mers are actually similar to each other, which makes running time really difficult to guess and this genome does look hardcore - it's huge and there is so much heterozygosity. I honestly would not try to run the pythonic version, it's not very efficient. tbenavi1/KMC is your best bet. It's still single threaded, but it's a lot faster (my guess would be ~3 days with your genome and a lot less memory, but it's just my guess).
What about if I would try to help you resolving the dependency problem instead?
Thank you for your quick response and apologies for my long delay. I am glad you've seen similarly complicated genomes! I agree that resolving the installation issues of tbenavi1/KMC will be important moving forward, but wanted to first update you that SmudgePlot completed on this assembly and surprisingly concluded that it is diploid! I've only just opened these so I haven't had much time to think deeply about what is known about this species (diploid, triploid and aneuploid lineages) and how these SmudgePlot results mesh with the results we have so far from the HiFiASM assembly and GenomeScope.
Based on the literature, we do expect the genome size between ~2.4-3.6 Gb depending on the lineage. Our assembly after HiFiASM was 5.17 Gb. We did an extra round of purging because we believed this to be a triploid specimen based on the what was known about the population that was collected as well as the GenomeScope results and achieved a genome size of 3.5 Gb. Given your experience, I'd love to hear any thoughts you might have on the diploid vs triploid nature of this assembly.
Following the method I included above, these plots were generated using these commands:
smudgeplot.py hetkmers -o kmer_counts_L"$L"_U"$U" < kmer_counts_L"$L"_U"$U".dump
smudgeplot.py plot -o Haemaphysalis_longicornis_HiFi -t "Haemaphysalis longicornis" kmer_counts_L"$L"_U"$U"_coverages.tsv
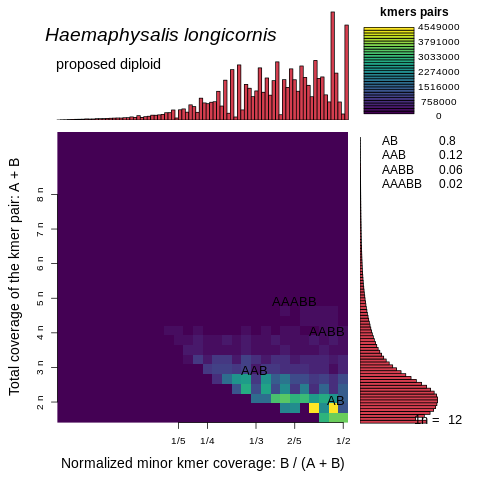
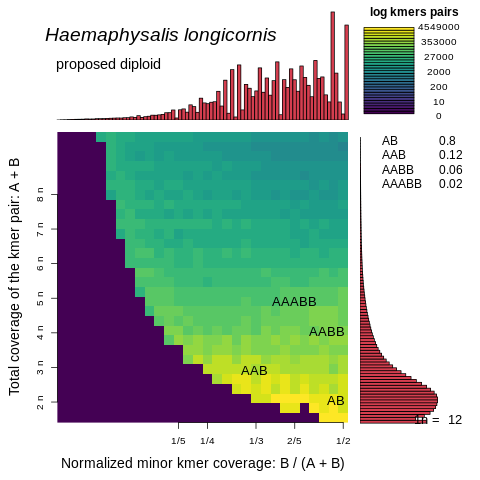
Thanks again for all your help so far!
A couple other things worth noting... purge_dups was unable to distinguish the peaks in coverage ([M::calcuts] Find 0 peaks) so I believe it only trimmed overlaps, but we are investigating it further as it looks like there were errors with the run.
The BUSCO completeness stats for the purged assembly were C:94.5%[S:85.3%,D:9.2%],F:2.3%,M:3.2%,n:2934. We are working on KAT and/or Merqury plots and aligning the Hi-C data.
Hello! Writing to update this thread with the latest results. I realized that purge_dups wasn't reading my alignments correctly, so had to re-run the whole thing.
Purge dups calculated cutoffs at -l 5 m -15 -h 45. Histogram attached here, which at least reflects the genomescope kmer histogram thankfully.
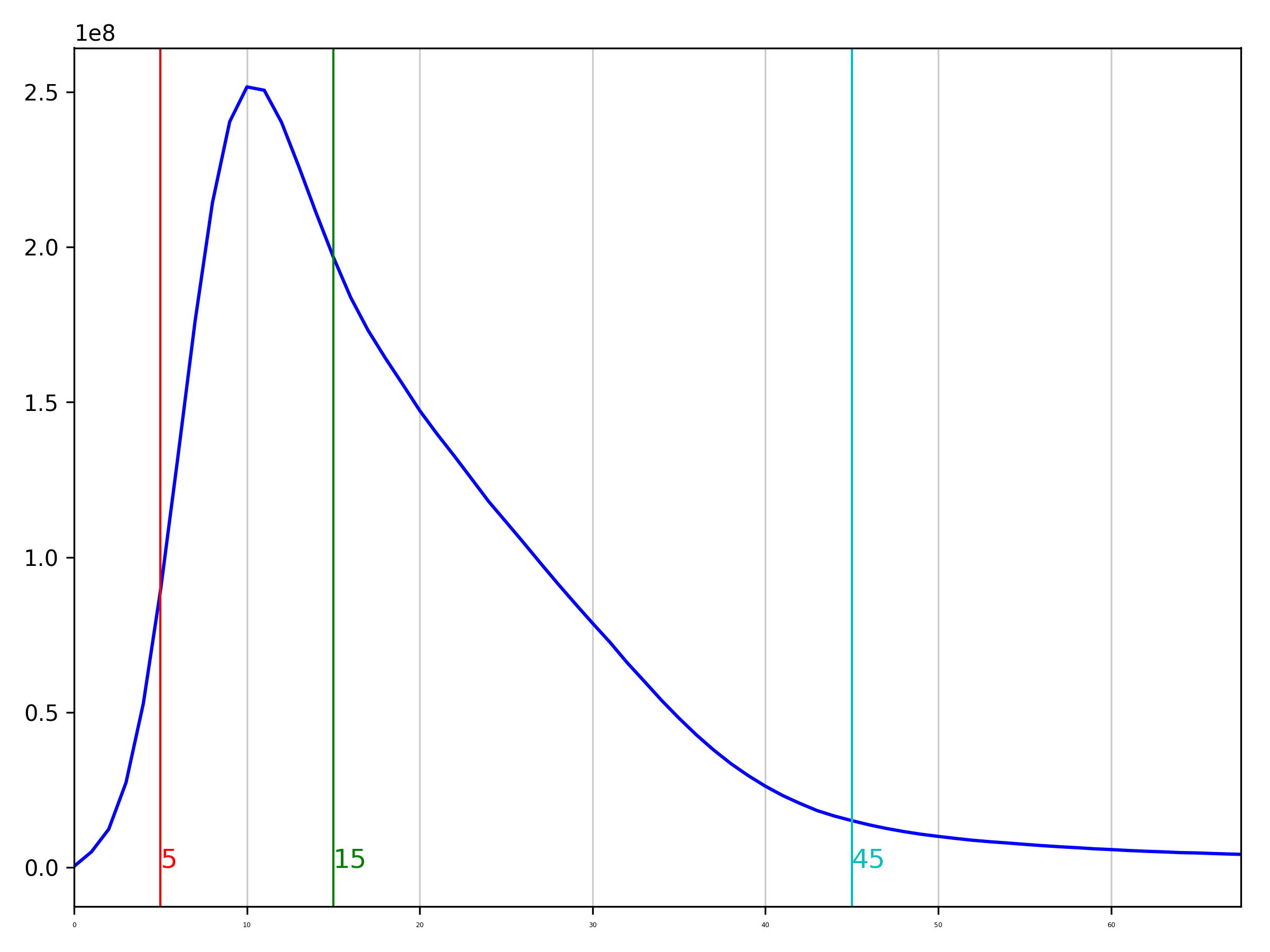
Using these cutoffs, purge_dups yielded an assembly with fewer duplicate arachnida BUSCOs but certainly still lots retained. It went from C:95.2%[S:29.0%,D:66.2%],F:2.0%,M:2.8% on the primary Hifiasm contigs to C:94.1%[S:73.1%,D:21.0%],F:2.2%,M:3.7% on the purged primary. In terms of total length, we went from from 5170.252 to 3560.207 MB.
Any advice to improve cutoffs on this to get it down to 1n assembly based on our smudgeplot results etc? I plan to also generate a couple of KAT kmer spectra plots from these assemblies. Anything else we should generate to get a better handle on what's happening here?
Thanks! Amanda
Wow, what a cheeky genome. I am not sure about the ploidy, the evidence is a bit of a mixed bag. The stuff I am quite convinced by: 1. the 1n kmer coverage is ~9.5 (for the k you used) 2. the two or three haplotypes are divergent from each other - it is both suggested by the high 1n peak and those really high BUSCO duplications values.
Our assembly after HiFiASM was 5.17 Gb. We did an extra round of purging because we believed this to be a triploid specimen based on the what was known about the population that was collected as well as the GenomeScope results and achieved a genome size of 3.5 Gb.
So, I am not 100% convinced "purging" is the way to go in the case. Everything indicates the haplotypes are quite divergent from each other, and therefore it might be worth constructing the full genome as it is. I don't think any ha/monoploid reference will be representative of the genome. KAT/Mercury is certainly a good idea to get an idea of what is in an assembly.
Did you annotate your genome? Any version would do, but the less purged the better. Does not have to be anything too fancy... But if you take the gene models and do a reciprocal blast, you could get a hint of how divergent the haplotypes are. That kind of stuff was done to understand better the root-knot nematode genome (e.g. this one), recently we did this to figure the origin of fungus gnat GRC chromosomes (https://doi.org/10.1371/journal.pbio.3001559) and we also have the code to do everything documented if you would be interested in pursuing this avenue.
Not sure if everything I write makes sense, perhaps at this point it might be easier to have a chat?
Hey @everyone, we have a working beta-version that is substantially faster and actually does the whole job a bit better; would you like to give it a try?
I will close this for now, though if you would like to try the beta-version, please do get in touch