Intron retention detection
Hello,
How does bambu take into account novel isoforms with intron retention?
I'm using human gencode.v41 annotations on hg38 genome and definitely have evidence of intron retention when looking at the bam files on IGV:
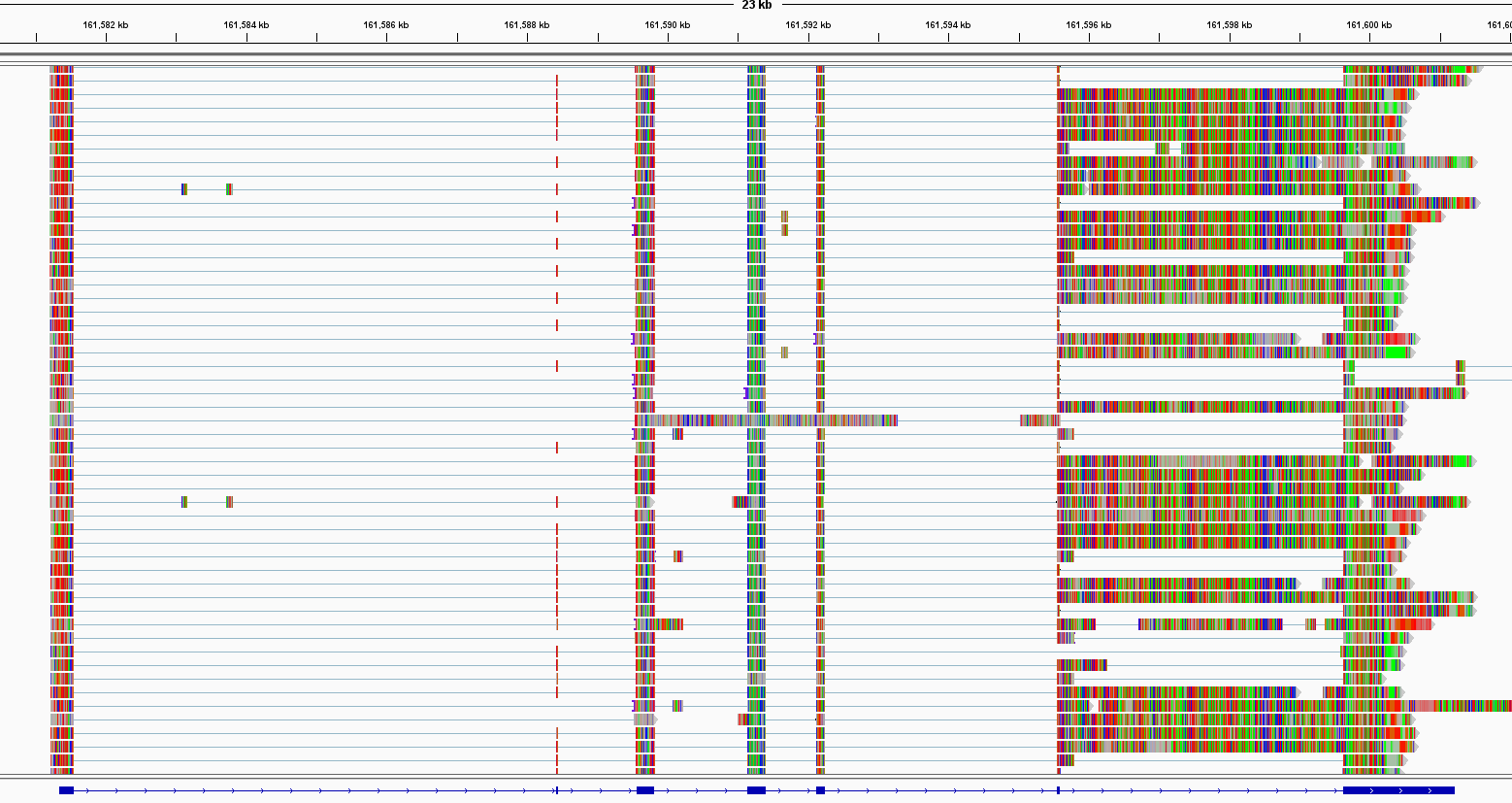
However, bambu doesn't identify any novel isoform for my gene of interest (7 exons), even if I increase the NDR to 0.3 or 0.5. I also tried to run the De novo transcript discovery mode.
Any help with this would be very much appreciated. Thank you!
Hi,
Bambu should treat novel intron retention transcripts like any other so long as as intron retention doesn't result in a single exon transcript, there are some niche exceptions.
In your example, are there any other annotations in this area? Specifically is there a transcript with all these exons, and not the last exon? I ask because Bambu groups reads together by exon-junctions, in this case the intron retention event fuses the last two exons together meaning it would be grouped together with the annotation not containing the final exon. The case of IR events including the final exon is particularly tricky because it could be either an alternate start or end site for an annotated, or an IR event, which is why Bambu groups them together for now.
Another possibility I just realized which is more likely since you also had this issue in de novo mode is that you can try run bambu with opt.discovery = list(remove.subsetTx=FALSE). By default bambu does not report subset transcripts because they have an extremely high enrichment in false positions. Because this transcript is "technically" a subset of the full transcript (because of the fused final exons means all its exon junctions are consecutively contained in another transcript) it is being filtered out by this filter.
Let me know if this is the case or not and I can try investigate further.
Hello Andre,
Thanks very much for your response.
Regarding your question "Specifically is there a transcript with all these exons, and not the last exon?" the answer is YES there is, although I don't think bambu is grouping the reads with intron retention with the ones with the last exon missing because the numbers don't add up.
I also tried the option you suggested using this command:
se <- bambu(reads = 'CRA125_mono_sorted.bam', annotations = bambuAnnotations, genome = 'GCA_000001405.15_GRCh38_no_alt_analysis_set.fa', ncore = 8, opt.discovery = list(remove.subsetTx=FALSE))
Is that the right way to do it because I end up with even less new isoforms than without the option and still no sign of my isoform with intron retention?
Hello again Andre,
A further (and related) issue we would like to understand is why bambu also seems to identify an upstream transcript when there is no evidence for it in our read alignments.
In the image below
-the top panel is the annotated transcripts for the gene of interest. According to bambu the transcript called ENST00000508651 (2C-210) makes up 40% of the reads mapping to that gene. However we see no evidence of that in IGV.
-the middle panel is the gtf annotation to see where the exons are located for that transcript
-the bottom panel is an IGV screenshot of the reads mapping to the gene of interest (FCGR2C) and we have roughly overlayed where the exons of 2C-210 should be with the double headed arrows across the bottom. You can see 1) no evidence of 2C-210 transcript 2) clearly evidence for retention of last intron
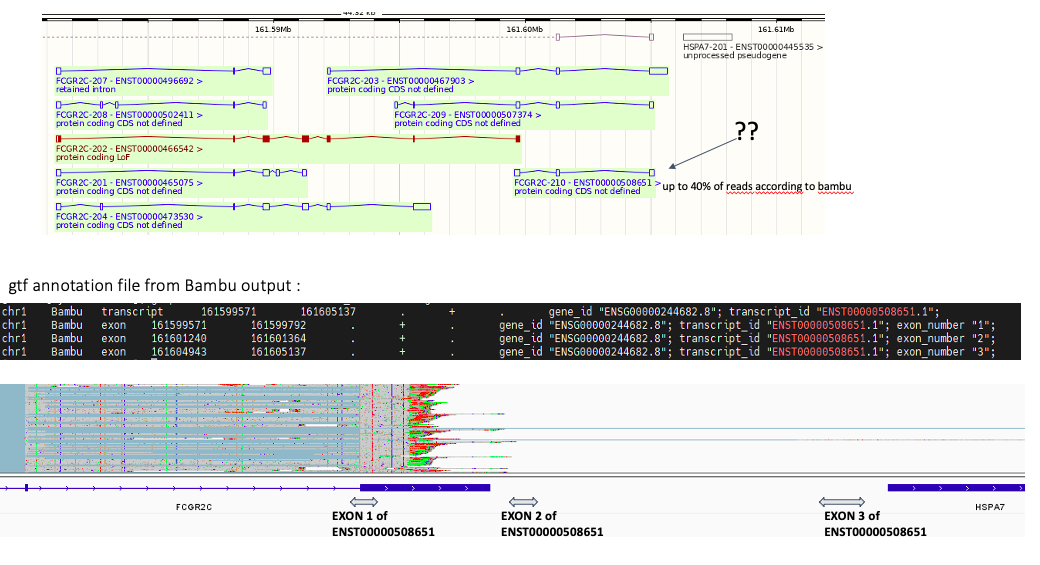
Perhaps these mistakes are related?
Thanks for your help!
Hi,
-
Existence of an annotation missing the last exon Ok just to clarify to make sure I understand what is going on here, what method are you using to get the counts? CPM? Full-length reads?
-
Running with subsets on results in fewer novel transcripts Yes this is expected behavior because of the impact the higher number of false positives allowing subsets introduce. Essentially you now increased the ratio of novel to annotated reads found in the sample. To solve this set a higher NDR when turning off the subset filter to get it to the number of novel transcripts you would like.
To save some time you can optionally can set quant = FALSE, set a output path with rcOutDir and run it with NDR = 1. You can then filter using the NDR in the mcols() of the annotation output. You can then pass this filtered annotation output into a second run of bambu (setting discovery = FALSE and passing the .rds file in your rcOutDir), to just do the quantification.
- Bambu quantifying a transcript with no evidence in IGV This is a tricky one. We have had an issue similar to this in the past where the issue ended up being in IGV which was sampling the reads out due to high sequencing depth. However if it represents 40% of the gene expression I doubt that is the case this time. I think I need more information to be able to solve/debug this one. If you run step 2. with the rcOutDir, are you able to send me the .rds file that is produced? Could you also send me the session info of running Bambu?
Thanks for using Bambu, I will do my best to try and solve this.