Inconsistent read counts within polycistronic transcripts with near-constant expression
In our mouse cDNA data, we hypothesise that mt-Atp6 and mt-Atp8 are encoded together on the mouse mitochondrial genome as a single polycistronic unit, which is demonstrated by observing co-expression at what appears to be essentially identical levels across the two genes in all samples:
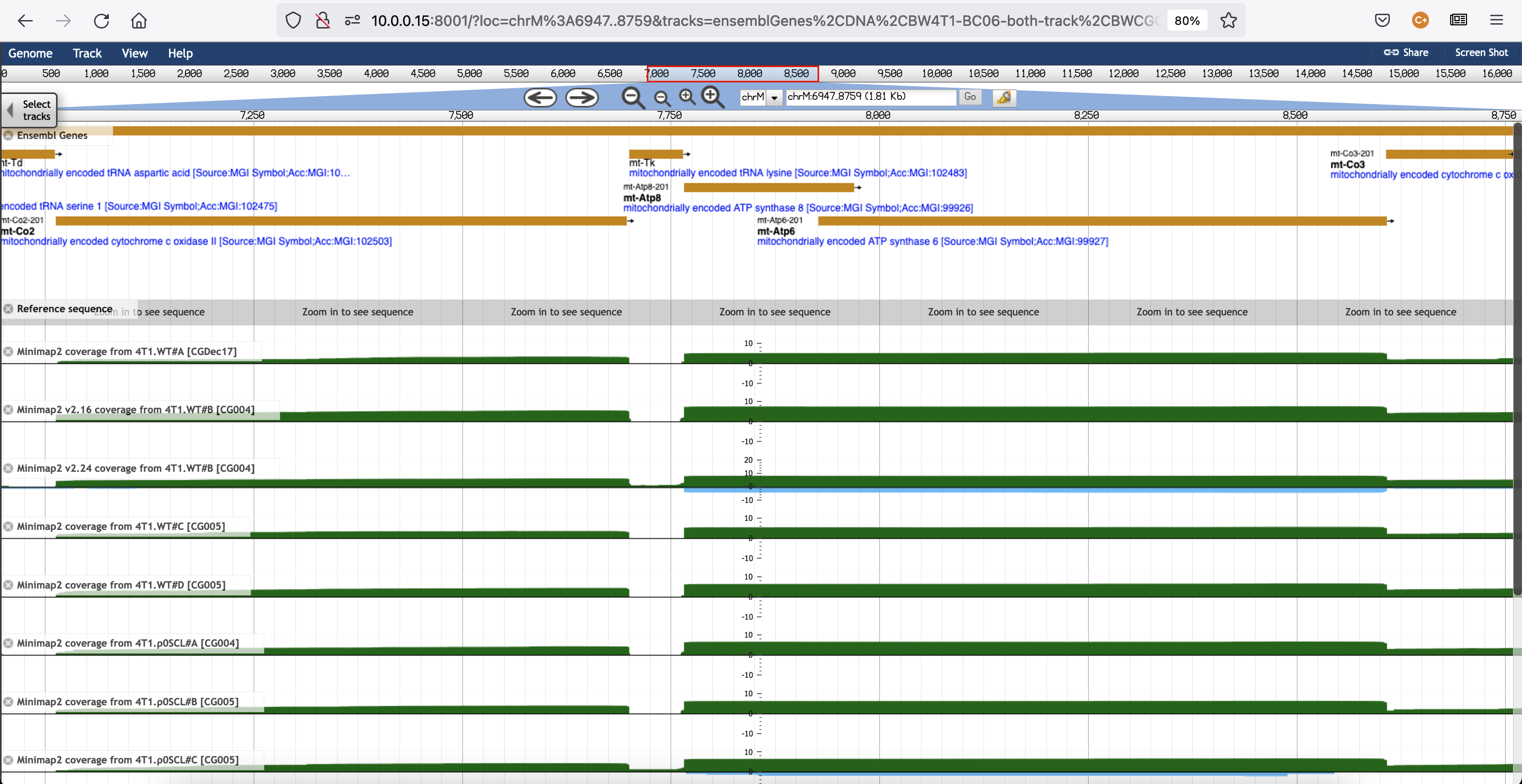
However, bambu has reported different counts for these transcripts, inconsistently different in different samples. This is most obviously demonstrated in the SCL samples shown below, with raw Atp6/Atp8 counts reported by bambu of 77/132, 155/64, and 115/3 for the three SCL samples. These counts should either be nearly identical (because expression levels across the region are the same), or counts should be counted for only one of the transcripts in the polycistronic unit in all cases. Identical expression would be preferred, but I can see where that could complicate matters if each cDNA read needs to be assigned to a single transcript. I suppose another alternative would be to create a fusion transcript composed of both annotated genes.

The polycistronic nature of this region may be easier to see in the SNP coverage track from JBrowse2 (different samples), showing that there is no blue polyA insertion point between the genes (as would be expected if the genes were encoded separately), and some of the mapped reads are mapping all the way through both genes:
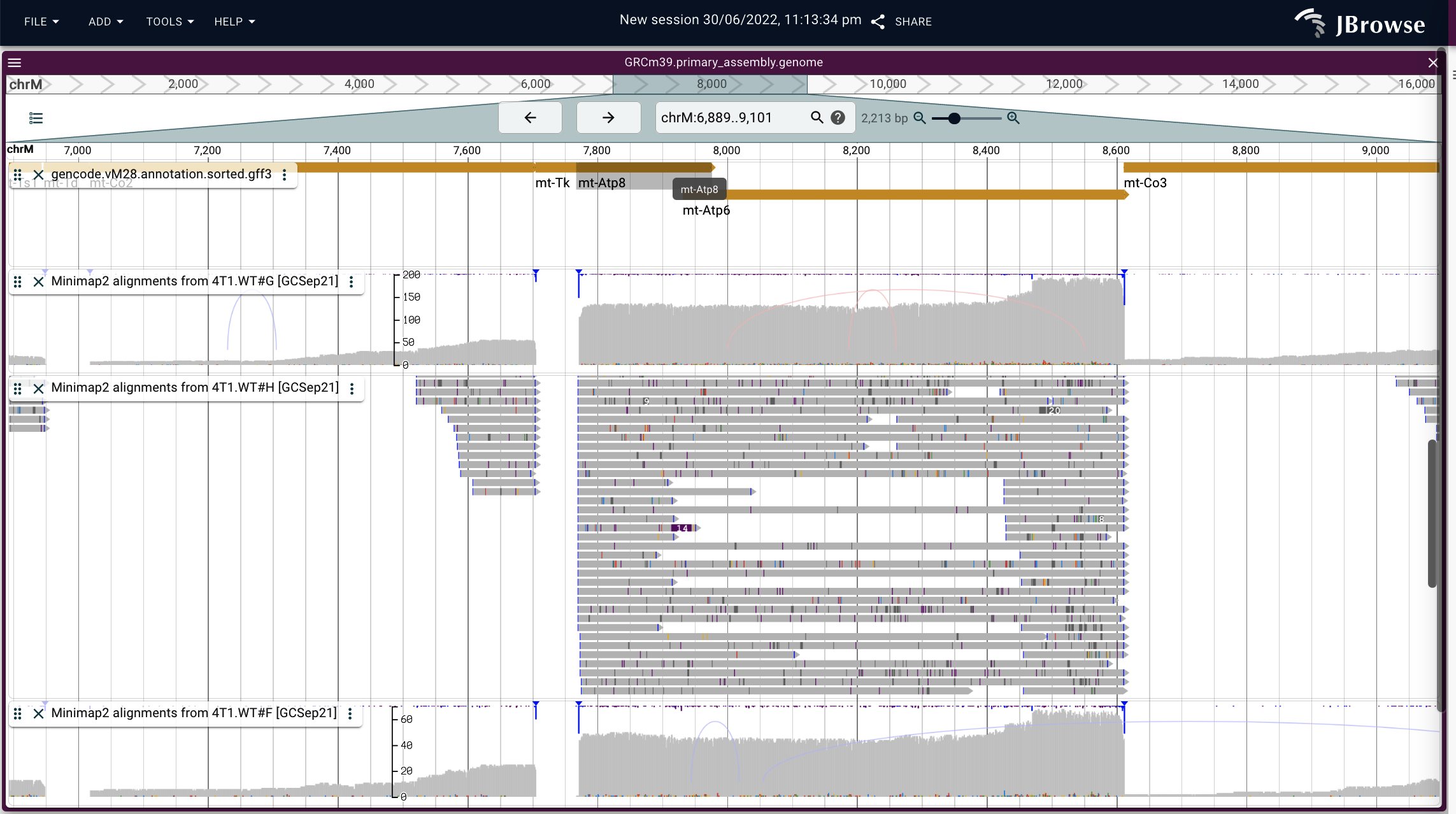
[the more fuzzy coverage in JBrowse2 is due to our version of JBrowse2 considering deletions in BAM CIGAR strings to be a drop in coverage, which means that the covered read count is inconsistent with the observed coverage]
I have included as an attachment a zip file containing subset BAM files for the Atp6/Atp8 region of the mitochondrial genome. Annotation/genome files are the same as for the previous issue [issue #281]. The associated .rds file (containing all discovered transcripts) can be found here.
This issue is possibly related to pull #260, which mentions multi-gene assignments and fusion transcripts.
Hi @gringer , this scenarios is a bit tricky. In fact, during estimation, for reads that map acorss multiple genes, they will usually be randomly assigned to one of the genes and currently we don't enforce any control on this, so it would very likely happen when different genes are chosen in different samples. Another possible reason that might explain this inconsistency could be there exist different unique read support for each of the atp7/atp8 genes, and that might also contribute to the inconsistency that you observed here. My suggestion for this case would be to construct a genome that contain this polycistronic transcript, and do alignment to this specific genome and then apply bambu for the quantification, it should then be able to give you the desired estimates.
Would it be possible to do something like flag reads that are multi-mapping, and assign their reads randomly at the end once all counts have been observed? I wouldn't mind if the reads were randomly assigned, but that's not the case here; there are clearly sample-specific systematic mapping differences for transcripts that (from what I can tell from the minimap2 coverage results) have essentially identical levels of expression. See the pink highlighted cells in the table screenshot.