Extreme mismatch between JBrowse coverage and Bambu mapping (moderate vs 0 expression)
After reporting Bambu results to the biologists I have been working with, they noticed a number of odd counting behaviours from the Bambu output.
One gene in particular that surprised me was Sdhc, a negative strand gene on chr1, ranked high in terms of its differential expression, but this differential expression was not represented in the transcripts.
This gene is obviously expressed in JBrowse in all samples:
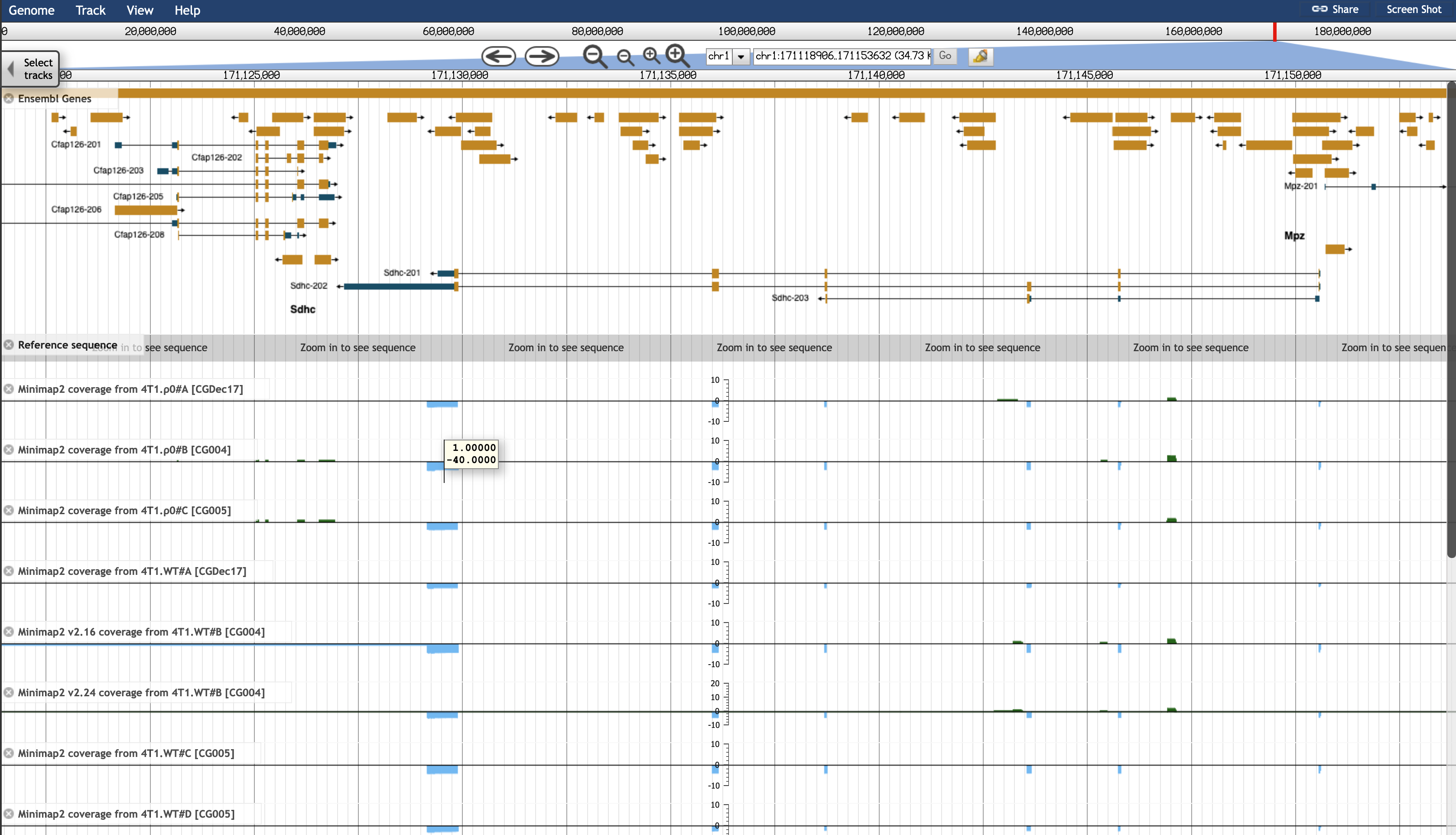
That JBrowse plot is from a mapping of an earlier basecalling of the reads to an earlier genome, but the situation is consistent with the BAM files that were interpreted by Bambu:
$ for x in Sdhc_*.bam; do echo -n "${x}: "; samtools idxstats ${x} | grep chr1[[:space:]]; done
Sdhc_mm2_DE_repeat_Jun22_called_p0_CGAug18_004_BC05_vs_genome.bam: chr1 195154279 113 0
Sdhc_mm2_DE_repeat_Jun22_called_p0_CGDec17_001_BC07_vs_genome.bam: chr1 195154279 29 0
Sdhc_mm2_DE_repeat_Jun22_called_p0_CGNov18_005_BC03_vs_genome.bam: chr1 195154279 45 0
Sdhc_mm2_DE_repeat_Jun22_called_SCL_CGNov18_005_BC04_vs_genome.bam: chr1 195154279 71 0
Sdhc_mm2_DE_repeat_Jun22_called_SCL_CGNov18_005_BC05_vs_genome.bam: chr1 195154279 54 0
Sdhc_mm2_DE_repeat_Jun22_called_SCL_CGNov18_005_BC06_vs_genome.bam: chr1 195154279 40 0
Sdhc_mm2_DE_repeat_Jun22_called_WT_CGAug18_004_BC04_vs_genome.bam: chr1 195154279 132 0
Sdhc_mm2_DE_repeat_Jun22_called_WT_CGDec17_001_BC06_vs_genome.bam: chr1 195154279 14 0
Sdhc_mm2_DE_repeat_Jun22_called_WT_CGNov18_005_BC01_vs_genome.bam: chr1 195154279 61 0
Sdhc_mm2_DE_repeat_Jun22_called_WT_CGNov18_005_BC02_vs_genome.bam: chr1 195154279 41 0
I ran bambu as follows:
library(bambu);
bam.files <- list.files("mapped", pattern=".*\\.bam$", full.names=TRUE);
names(bam.files) <- sub("^.*_called_(.*)_vs_.*$", "\\1", bam.files);
bam.fileList <- Rsamtools::BamFileList(bam.files);
fa.file <- "~/db/fasta/mmus/gencode/M28/GRCm39.primary_assembly.genome.fa";
gtf.file <- "~/db/fasta/mmus/gencode/M28/gencode.vM28.primary_assembly.annotation.gtf";
bambuAnnotations <- prepareAnnotations(gtf.file);
se <- bambu(reads = bam.fileList, annotations = bambuAnnotations, genome = fa.file, verbose=TRUE, discovery=FALSE, stranded=TRUE, ncore=10);
Despite this coverage, bambu reports zero counts in some, but not all, samples:
se.disc <- readRDS("bambu_WT_vs_p0_GenCode_M28_withDiscovery.rds");
## Remove version number from transcript
rowData(se.disc)$TXNAME <- sub("(ENSMUST.*?)\\..*$","\\1",rowData(se.disc)$TXNAME);
> assays(se.disc)$counts[rowData(se.disc)$TXNAME %in% (filter(tsyms.tbl, name == "Sdhc") %>% pull(transcript)),]
p0_CGAug18_004_BC05 p0_CGDec17_001_BC07 p0_CGNov18_005_BC03 SCL_CGNov18_005_BC04
ENSMUST00000081560.5 0 0 0 1
ENSMUST00000111336.10 0 0 0 63
ENSMUST00000155798.2 0 0 0 0
SCL_CGNov18_005_BC05 SCL_CGNov18_005_BC06 WT_CGAug18_004_BC04 WT_CGDec17_001_BC06
ENSMUST00000081560.5 2.390495e-08 0 4.274021e-09 1
ENSMUST00000111336.10 3.900000e+01 0 1.040000e+02 13
ENSMUST00000155798.2 0.000000e+00 0 0.000000e+00 0
WT_CGNov18_005_BC01 WT_CGNov18_005_BC02
ENSMUST00000081560.5 1 1
ENSMUST00000111336.10 52 24
ENSMUST00000155798.2 0 0
There are other odd genes in the data set; this is just the one that I think is the most obviously wrong. What I find most interesting is that bambu has expression in two of the three sample types (WT, SCL), but not the other one, suggesting that it's found an appropriate matching gene model, but for some reason doesn't consider it appropriate for the p0 cell lines.
I've attached data files to this issue:
- the SummarizedExperiment that bambu produced [
bambu_WT_vs_p0_GenCode_M28_withDiscovery.rds] - ten BAM files from my reads, filtered on the Sdhc gene region [
Sdhc_mm2_DE_repeat_Jun22_called_<SampleType>_<SampleID>_vs_genome.bam]
I mapped these reads to the Gencode M28 genome [GRCm39.primary_assembly.genome.fa and gencode.vM28.primary_assembly.annotation.gtf from here]
Just in case it matters...
> sessionInfo()
R version 4.1.2 (2021-11-01)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Debian GNU/Linux bookworm/sid
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.19.so
locale:
[1] LC_CTYPE=en_NZ.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_NZ.UTF-8 LC_COLLATE=en_NZ.UTF-8
[5] LC_MONETARY=en_NZ.UTF-8 LC_MESSAGES=en_NZ.UTF-8
[7] LC_PAPER=en_NZ.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_NZ.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] bambu_2.0.6 BSgenome_1.62.0
[3] rtracklayer_1.54.0 Biostrings_2.62.0
[5] XVector_0.34.0 SummarizedExperiment_1.24.0
[7] Biobase_2.54.0 GenomicRanges_1.46.1
[9] GenomeInfoDb_1.30.0 IRanges_2.28.0
[11] S4Vectors_0.32.3 BiocGenerics_0.40.0
[13] MatrixGenerics_1.6.0 matrixStats_0.61.0
loaded via a namespace (and not attached):
[1] Rcpp_1.0.8 lattice_0.20-45 tidyr_1.1.4
[4] prettyunits_1.1.1 png_0.1-7 Rsamtools_2.10.0
[7] assertthat_0.2.1 digest_0.6.29 utf8_1.2.2
[10] BiocFileCache_2.2.1 R6_2.5.1 RSQLite_2.2.9
[13] httr_1.4.2 pillar_1.6.5 zlibbioc_1.40.0
[16] rlang_1.0.0 GenomicFeatures_1.46.5 progress_1.2.2
[19] curl_4.3.2 data.table_1.14.2 blob_1.2.2
[22] Matrix_1.4-0 BiocParallel_1.28.3 stringr_1.4.0
[25] RCurl_1.98-1.5 bit_4.0.4 biomaRt_2.50.3
[28] DelayedArray_0.20.0 compiler_4.1.2 pkgconfig_2.0.3
[31] tidyselect_1.1.1 KEGGREST_1.34.0 tibble_3.1.6
[34] GenomeInfoDbData_1.2.7 XML_3.99-0.8 fansi_1.0.2
[37] crayon_1.4.2 dplyr_1.0.7 dbplyr_2.1.1
[40] rappdirs_0.3.3 GenomicAlignments_1.30.0 bitops_1.0-7
[43] grid_4.1.2 jsonlite_1.7.3 lifecycle_1.0.1
[46] DBI_1.1.2 magrittr_2.0.1 cli_3.1.1
[49] stringi_1.7.6 cachem_1.0.6 xml2_1.3.3
[52] filelock_1.0.2 ellipsis_0.3.2 vctrs_0.3.8
[55] generics_0.1.1 xgboost_1.6.0.1 rjson_0.2.21
[58] restfulr_0.0.15 tools_4.1.2 bit64_4.0.5
[61] glue_1.6.1 purrr_0.3.4 hms_1.1.1
[64] parallel_4.1.2 fastmap_1.1.0 yaml_2.2.2
[67] AnnotationDbi_1.56.2 memoise_2.0.1 BiocIO_1.4.0
Edit: got my R system wrong for sessionInfo; I'm running Bambu remotely, then running DESeq2 on a different system.
Hi @gringer , this was caused by one of the bug which we have now fixed in a separate branch that you may use (https://github.com/GoekeLab/bambu/tree/modifyQuant), and with this branch, it should report the values as expected.
Do let us know if there are more questions regarding the quantification results.
Thank you
Thanks, we've got other weird results, so I'll check out that branch and then see how many of the other results it fixes.
Just confirming that this has fixed the immediate problem we have been having with Sdhc counts:
>assays(se.disc)$counts[rowData(se.disc)$TXNAME %in% (filter(tsyms.tbl, name == "Sdhc") %>% pull(transcript)),]
p0_CGAug18_004_BC05 p0_CGDec17_001_BC07 p0_CGNov18_005_BC03 SCL_CGNov18_005_BC04
ENSMUST00000081560.5 0 0 0 0
ENSMUST00000111336.10 78 19 31 59
ENSMUST00000155798.2 0 0 0 0
SCL_CGNov18_005_BC05 SCL_CGNov18_005_BC06 WT_CGAug18_004_BC04 WT_CGDec17_001_BC06
ENSMUST00000081560.5 3.455636e-08 0 4.274021e-09 1
ENSMUST00000111336.10 3.700000e+01 31 1.040000e+02 13
ENSMUST00000155798.2 0.000000e+00 0 0.000000e+00 0
WT_CGNov18_005_BC01 WT_CGNov18_005_BC02 p0_CGAug18_004_BC05 p0_CGDec17_001_BC07
ENSMUST00000081560.5 1 0 0 0
ENSMUST00000111336.10 49 23 78 19
ENSMUST00000155798.2 0 0 0 0
p0_CGNov18_005_BC03 SCL_CGNov18_005_BC04 SCL_CGNov18_005_BC05 SCL_CGNov18_005_BC06
ENSMUST00000081560.5 0 0 3.455636e-08 0
ENSMUST00000111336.10 31 59 3.700000e+01 31
ENSMUST00000155798.2 0 0 0.000000e+00 0
WT_CGAug18_004_BC04 WT_CGDec17_001_BC06 WT_CGNov18_005_BC01 WT_CGNov18_005_BC02
ENSMUST00000081560.5 4.274021e-09 1 1 0
ENSMUST00000111336.10 1.040000e+02 13 49 23
ENSMUST00000155798.2 0.000000e+00 0 0 0