Associating read IDs with transcript IDs
Hello, I was wondering if there is a way to associate transcript IDs to their respective read IDs, so that one would be able to subset the bam file to only keep the reads that map to a specific transcript (or a list of transcripts)?
Thank you!
Hi, We do have this as a beta feature in the read_to_model_map branch. With this version Bambu needs to be run with trackReads = TRUE and will add a metadata(se)$readModelMap to the output from the bambu function if running with quantification. If you run it in discovery mode only, it will instead output a list containing both the annotations and the read model map.
Unfortunately this branch is using a slightly older version of Bambu so your results may differ depending on your data. I will look in the coming days if I can merge it with the latest release, but I will need some time for this.
I hope this helps and let me know how it goes.
Hi, Thanks for your answer. I will see if I can make it work; otherwise I will wait to see if you can merge the branch with the latest release :)
Thanks!
Hi there, I have updated the BETA read_to_model_map branch to the latest release. Please let me know if you have any success with it.
Hello,
Thanks a lot for the update! Could you let me know how I can use this though, I'm not sure what the command is and what output I can get from there? Once I have the se object, where do I go from there? Thanks in advance.
Sure, you run bambu as normal but add trackReads = TRUE
se <- bambu(reads = test.bam, annotations = bambuAnnotations, genome = fa.file, trackReads = TRUE)
You can access the map between the reads and the transcript models via
metadata(se)$readModelMap
This is a matrix with the first column being the read ids, and the second being the transcript ids. From there you should be able to filter this matrix to get only the read ids that belong to your transcript ids of interest. Keep in mind this only associates full length reads in a one to one relationship (i.e reads that could be used to construct the transcript model), partially mapped reads (for example those that map to only a single exon of the transcript are not mapped) will not be linked with their larger transcript
Hello, I'm sorry for not answering earlier, I will give this a go during the next week and will let you know how it goes! Thanks a lot for all the help.
Hello,
I tried running the command you wrote with bambu I installed with bioconductor a month ago, but got the following message:
Error in bambu(reads = bamFiles, annotations = bambuAnnotations, genome = genomeSequence, : unused argument (trackReads = TRUE)
Do I have to somehow update it or install a different version?
Thank you very much.
Hi,
The ability to track read ID's to the transcript IDs is not yet in the bioconductor version as it is still a beta feature. You need to download the read_to_model_map branch from github and use that version.
In bash
git clone --branch read_to_model_map https://github.com/GoekeLab/bambu.git
In R
library(devtools)
load_all("path/to/bambu")
Then you can try run bambu again.
Hope this helps.
Hello,
Thanks a lot for all the help!
I managed to get the table with read IDs and transcript IDs, eg.:
"ad1f43c8-e4d4-4580-bde6-eb07a51f2c6d" "ENSMUST00000029546.15"
Let's see if I can make sense of it now :)
Again thanks a lot, I think this feature can be very useful :)
Cheers, Ivan
Hello,
So I looked into individual transcript IDs, and some of the transcripts that were reported in the bambu output were not present in this table. Based on your explanation above, this means that the reads mapping to these transcripts were not mapping to all of the exons present in the transcript, but only some of them, and therefore they are not reported in the readID-transcriptID table?
Here is an example: tx.5827 is a highly expressed transcript in all 3 samples I analyzed. When I search for it in the counts_transcript.txt file, this is what I get:
grep tx.5825 counts_transcript.txt
TXNAME GENEID Sample1.bam Sample2.bam Sample3.bam
tx.5827 ENSMUSG00000068823.11 71.2933765465939 63.1532703901375 75.7576378802568
but then when I search for it in the readID-transcriptID table:
grep tx.5827 readID_transcriptID_withDiscovery.csv
145831,2882ceb3-bde4-4ad3-893d-1a1c2083cc62,tx.5827
145832,ad69a368-d3e7-42d5-8f1d-113625b6e729,tx.5827
so only 2 reads are reported to map to this transcript.
Is there a way I could somehow still find all the missing reads and find out where they were mapping to?
Thanks a lot in advance.
Hi Ivan,
Glad you were able to get this working.
Could you please check for me how many full length reads were assigned to tx.5827
assays(se)$fullLengthCounts["tx.5827",]
This will help be get an idea if its Bambu that is making a mistake or if these gene only has 2 full length reads.
Another helpful way to see where these missing reads are would be to load your bam file in a genome browser (like IGV) and you can zoom into your gene of interest and it will show the reads that are mapping. If you do manage to do that and it disagrees with the readID-transcriptID table, sending me a picture of this region would be very appreciated so I can try and solve the problem.
Hi Andre,
So indeed there are only 2 full length reads:
assays(se_readID_with_discovery)$fullLengthCounts["tx.5827",]
Sample1.bam Sample2.bam Sample2.bam
2 0 0
Here is a snapshot of the reads in IGV.
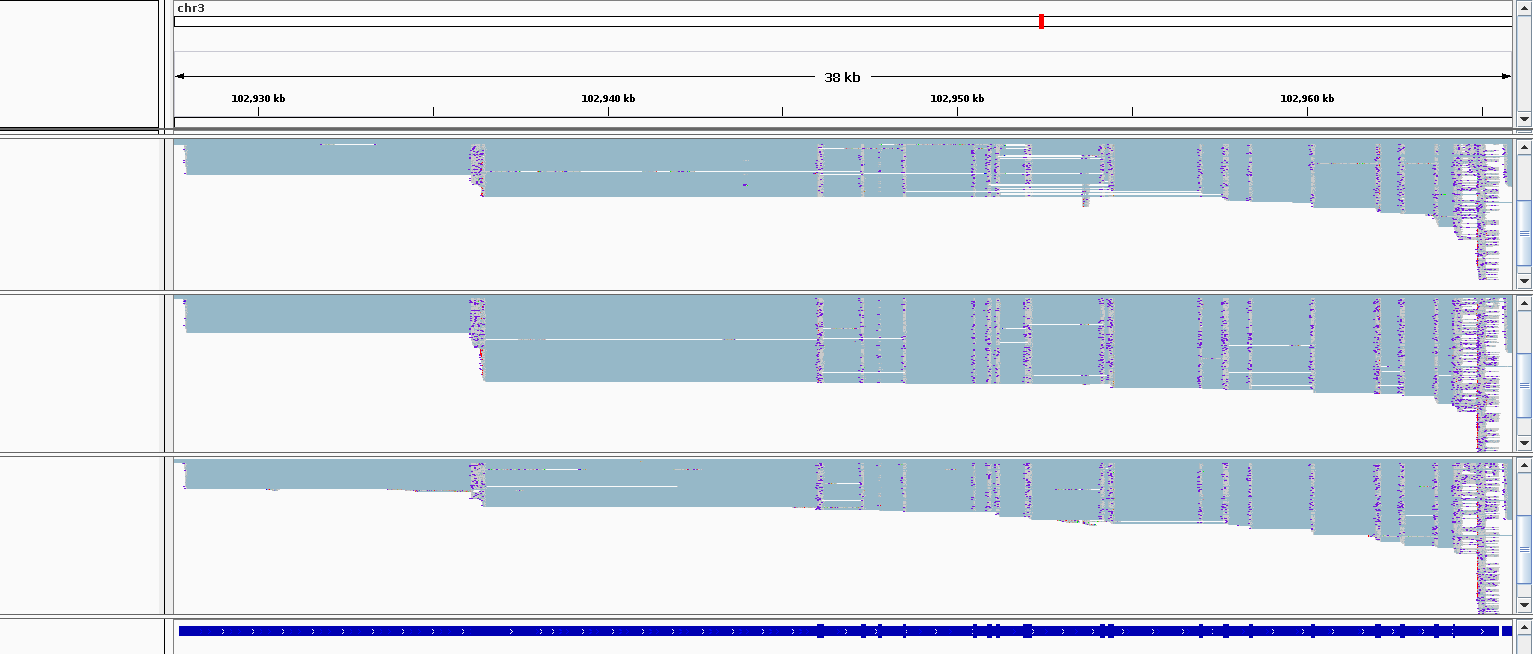
Is there any other way I can track the faith of non-full length reads? :)
Thanks!
Hi,
It does look like there are a lot of reads in this region so I am surprised only two would be classified as full length. Is it possible to expand out these annotations and the reads for one of the bams (right click on the track change change it from compressed) so I can see exactly which exons tx.5827 has. Just so I can confirm if this is a bug with Bambu or expected behavior.
We don't support partial matching reads to transcript ids as it gets complicated as partial matches could then map to multiple transcripts. However I can think of one way you could do this using Bambu in an unintended way (I can't guarantee it will work well and can't provide much support).
- Run Bambu again, increasing the NDR parameter to 1. What this does it treat all partial reads as a novel discovered transcripts and therefore they would be classed as full length reads.
- Output your new novel transcripts to a gtf file
writeToGTF(rowRanges(se))and load this track into IGV - Look for the novel transcript annotations that partially match tx.5827
- Uses these new transcript ids alongside tx.5827 to search for the reads in the new readID_transcriptID_withDiscovery.csv that you produce with an NDR of 1.
Hi, be on the look out for the next release of bambu as we have added partial matches to the trackReads output. See the documentation in the github readme for more information.