whether to use pipeline D or additional pipeline C when protein data exists?
My genome is small (180 Mb) and haploid. As BRAKER2 evidence I have good mapped RNA seq data from this organism, and ~550 same-species/close-species protein sequences.
Formerly I would use additional pipeline C ["training GeneMark-ET on the basis of RNA-Seq spliced alignment information, training AUGUSTUS on a set of training gene structures compiled from RNA-Seq supported gene structures predicted by GeneMark-ET and spliced alignment of proteins of a very closely related species"], but now I see "This pipeline is deprecated since pipeline D can also use proteins of closely related species in addition to OrthoDB." (Ditto additional pipeline A, which uses close-protein data for Augustus prediction but not training)
Pipeline D and additional pipelines A/C seem quite different (e.g. OrthoDB/ProtHint/GeneMarkETP+ vs. proteins.fa/genomethreader/GenemarkET). Closely-related protein seqs can be added to OrthoDB input for pipeline D.
However the warning for pipeline D is "we found this approach to work well for large genomes, but accuracy on small and medium sized genomes is unstable."
So I'm trying to decide which approach is best -- pipeline D, because my protein data set is limited to 550 sequences. and OrthoDB input could greatly augment that; but arguing against pipeline D, my genome is too small -- or stay with additional pipelines A/C?
(Or, not use protein evidence at all, since that seems to prevent BRAKER from predicting UTRs??)
Hello, I would recommend using the pipeline D (and adding OrthoDB proteins to your closely related ones). You can still add UTRs to the pipeline D, as illustrated in https://github.com/Gaius-Augustus/BRAKER#testing-braker-with-proteins-of-any-evolutionary-distance-and-rna-seq.
The meaning of accuracy on small and medium-sized genomes is unstable is a bit misleading. What be observed is that in some genomes, it was better to use proteins alone rather than proteins + RNA-Seq. This was true especially when there were many close proteins available, I would not worry about that since you do not have many closely related proteins.
Hope this helps, Tomas
At your suggestion I am currently running pipeline D, using the protozoan OrthoDB data set as protein input plus 23 sequence additions from me -- total # of sequences =1,774,003
braker.pl \
--genome=Apr20_softmasked.fasta \
--etpmode \
--prot_seq=my_protein_evidence.fasta \
--bam=4star_2pass_May4Aligned.out.bam \
--softmasking \
--cores=$SLURM_CPUS_PER_TASK \
--workingdir=braker2_May5_pipelineD_1 \
--gff3
I allotted this amount of resources:
#SBATCH --cpus-per-task=10 #SBATCH --mem=10GB
NB "4star_2pass_May4Aligned.out.bam" was generated by STAR with these parameters below, which are intended to 1) output bam files (sorted and unsorted), 2) add an intron flag to the bam files, and 3)reduce false introns . 4) 2pass mode is supposed to give better results when not using annotated input. The output file 4star_2pass_May4Aligned.out.bam is Unsorted, and is the one used in the BRAKER run above.
STAR --genomeDir genome_idx2a \
--readFilesIn SDW_G3_all_R1.fastq SDW_G3_all_R2.fastq \
--outSAMtype BAM Unsorted SortedByCoordinate \ #1
--outFileNamePrefix 4star_2pass_May4 \
--runThreadN 8 \
--outSAMstrandField intronMotif \ #2
--twopassMode Basic \ #4
--outFilterType BySJout \ #3
--alignSJoverhangMin 8 #3
The BRAKER pipeline has been running for nearly 2 days now but has been stuck here since day 1:
# Wed May 5 19:48:45 2021: starting prothint.py
/opt/ProtHint-2.6.0/bin/prothint.py --threads=10 --geneMarkGtf /braker2_May5_pipelineD_1/GeneMark-ES/genemark.gtf /braker2_May5_pipelineD_1/genome.fa /braker2_May5_pipelineD_1/proteins.fa
perhaps you can advise? It is allotted 7 more days to run, if needed. (But I feel it should not be taking so long...)
The RNA-Seq alignment looks good to me
The BRAKER pipeline has been running for nearly 2 days now but has been stuck here since day 1:
ProtHint outputs its progress to stderr, please check that to see at what stage it is.
The only error files generated as of now ( the script is still running) are these:
[sullis02@log-1 braker2_May5_pipelineD_1]$ find . -name "*err"
./errors/find_python3_re.err
./errors/new_species.stderr
./errors/GeneMark-ES.stderr
./errors/find_python3_biopython.err
None of these prothint outputs, I think? All of them are empty files.
The dirs and files generated so far are:
[sullis02@log-1 braker2_May5_pipelineD_1]$ ls -lgF --group-directories-first | cut -d " " -f 4-15
4096 May 5 19:49 diamond/
4096 May 5 18:55 errors/
4096 May 5 19:48 GeneMark-ES/
4096 May 5 18:55 GeneMark-ETP/
262144 May 7 01:23 Spaln/
4096 May 5 18:55 species/
9897 May 5 19:48 braker.log
513 May 5 19:48 cmd.log
5719 May 5 19:46 GeneMark-ES.stdout
4119013 May 5 19:49 gene_stat.yaml
183740633 May 5 18:55 genome.fa
9416 May 5 18:55 genome_header.map
224511842 May 5 19:51 nuc.fasta
907491928 May 5 18:55 proteins.fa
907491620 May 5 19:48 protwu9c4wen
19407749 May 5 19:49 seed_proteins.faa
1640 May 5 18:55 what-to-cite.txt
The last lines of braker.log are still
[# Wed May 5 19:48:45 2021: starting prothint.py
/opt/ProtHint-2.6.0/bin/prothint.py --threads=10 --geneMarkGtf /braker2_May5_pipelineD_1/GeneMark-ES/genemark.gtf /braker2_May5_pipelineD_1/genome.fa /braker2_May5_pipelineD_1/proteins.fa
The only files generated/changed since May 5 are in Spaln/
148278619 May 7 01:23 spaln.regions.gff
0 May 6 12:44 nuc_314041_prot_314041
8012 May 6 12:44 batch_3141_out
2300 May 6 12:43 batch_3141
564 May 6 12:43 prot_314100
6530 May 6 12:43 nuc_314100
564 May 6 12:43 prot_314099
6530 May 6 12:43 nuc_314099
(etc)
i.e., no new output has been generated anywhere since spaln.regions.gff
I notice the slurm file for this batch process contain ProtHint run logging. I am attaching it here
Hello,
it looks like ProtHint is stuck at 99.9%. My best guess is that this error occurred https://github.com/gatech-genemark/ProtHint/issues/14.
Do you have access to the node the code is running on? Killing the stuck Spaln process should immediately solve this problem. If not, let me know -- I wanted to fix this for a while, I can try to make a fix tomorrow and send you a version that will have a timeout for hanging Spaln alignments.
Tomas
Based on your suggestios our sysadmin killed process 2281719
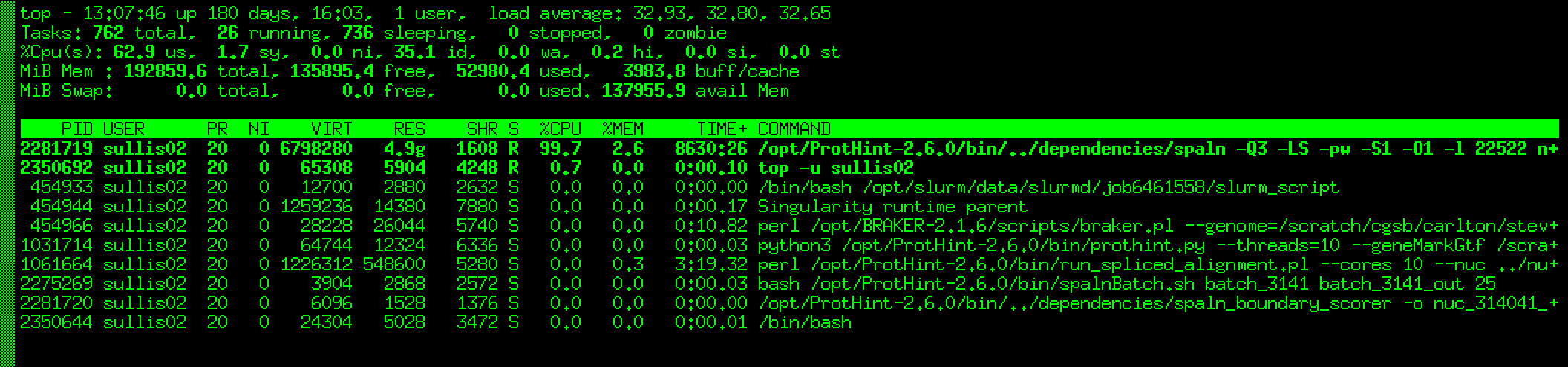
This immediately produced an output file tmp and another much larger file Spaln/spaln.gff, and then the whole BRAKER pipeline died without finishing.
tmp contains lines like this
]$ head tmp
Contig1_pilon_pilon Spaln_scorer start_codon 1924117 1924119 . + 0 prot=412133_1:015690; al_score=0.487603; eScore=5360; seed_gene_id=8989_g; topProt=TRUE;
Contig1_pilon_pilon Spaln_scorer CDS 1924117 1927239 . + 0 prot=412133_1:015690; exon_id=1; eScore=5360; seed_gene_id=8989_g; topProt=TRUE;
Contig1_pilon_pilon Spaln_scorer stop_codon 1927240 1927242 . + 0 prot=412133_1:015690; al_score=0.512397; eScore=5360; seed_gene_id=8989_g; topProt=TRUE;
Contig4_pilon_pilon Spaln_scorer start_codon 13650408 13650410 . + 0 prot=412133_1:00abf7; al_score=0.461157; eScore=991; seed_gene_id=31375_g; topProt=TRUE;
Contig4_pilon_pilon Spaln_scorer CDS 13650408 13650989 . + 0 prot=412133_1:00abf7; exon_id=1; eScore=991; seed_gene_id=31375_g; topProt=TRUE;
Contig4_pilon_pilon Spaln_scorer stop_codon 13650990 13650992 . + 0 prot=412133_1:00abf7; al_score=0.568595; eScore=991; seed_gene_id=31375_g; topProt=TRUE;
Contig4_pilon_pilon Spaln_scorer start_codon 13650408 13650410 . + 0 prot=412133_1:00ddf7; al_score=0.31405; eScore=610; seed_gene_id=31375_g;
Contig4_pilon_pilon Spaln_scorer CDS 13650408 13650939 . + 0 prot=412133_1:00ddf7; exon_id=1; eScore=610; seed_gene_id=31375_g;
Contig4_pilon_pilon Spaln_scorer start_codon 13650408 13650410 . + 0 prot=1144522_1:003e64; al_score=0.0380165; eScore=375; seed_gene_id=31375_g;
Contig4_pilon_pilon Spaln_scorer CDS 13650408 13650890 . + 0 prot=1144522_1:003e64; exon_id=1; eScore=375; seed_gene_id=31375_g;
And Spaln/spaln.gff
$ head Spaln/spaln.gff
Contig1_pilon_pilon Spaln_scorer start_codon 1924117 1924119 . + 0 prot=412133_1:015690; al_score=0.487603; eScore=5360; seed_gene_id=8989_g;
Contig1_pilon_pilon Spaln_scorer CDS 1924117 1927239 . + 0 prot=412133_1:015690; exon_id=1; eScore=5360; seed_gene_id=8989_g;
Contig1_pilon_pilon Spaln_scorer stop_codon 1927240 1927242 . + 0 prot=412133_1:015690; al_score=0.512397; eScore=5360; seed_gene_id=8989_g;
Contig4_pilon_pilon Spaln_scorer start_codon 13650408 13650410 . + 0 prot=412133_1:00abf7; al_score=0.461157; eScore=991; seed_gene_id=31375_g;
Contig4_pilon_pilon Spaln_scorer CDS 13650408 13650989 . + 0 prot=412133_1:00abf7; exon_id=1; eScore=991; seed_gene_id=31375_g;
Contig4_pilon_pilon Spaln_scorer stop_codon 13650990 13650992 . + 0 prot=412133_1:00abf7; al_score=0.568595; eScore=991; seed_gene_id=31375_g;
Contig4_pilon_pilon Spaln_scorer start_codon 13650408 13650410 . + 0 prot=412133_1:00ddf7; al_score=0.31405; eScore=610; seed_gene_id=31375_g;
Contig4_pilon_pilon Spaln_scorer CDS 13650408 13650939 . + 0 prot=412133_1:00ddf7; exon_id=1; eScore=610; seed_gene_id=31375_g;
Contig4_pilon_pilon Spaln_scorer start_codon 13650408 13650410 . + 0 prot=1144522_1:003e64; al_score=0.0380165; eScore=375; seed_gene_id=31375_g;
Contig4_pilon_pilon Spaln_scorer CDS 13650408 13650890 . + 0 prot=1144522_1:003e64; exon_id=1; eScore=375; seed_gene_id=31375_g;
$ wc -l tmp
358612 tmp
$ wc -l Spaln/spaln.gff
1563277 Spaln/spaln.gff
The final action comment in braker.log is still
# Wed May 5 19:48:45 2021: starting prothint.py
The final slurm lines are
[Fri May 7 01:22:54 2021] Enqueueing pair 613722/616186 (99.6%). Est. time left: 00:07:07 (hh:mm:ss)
[Fri May 7 01:23:02 2021] Enqueueing pair 614338/616186 (99.7%). Est. time left: 00:05:20 (hh:mm:ss)
[Fri May 7 01:23:09 2021] Enqueueing pair 614954/616186 (99.8%). Est. time left: 00:03:34 (hh:mm:ss)
[Fri May 7 01:23:13 2021] Enqueueing pair 615570/616186 (99.9%). Est. time left: 00:01:47 (hh:mm:ss)
[Wed May 12 13:08:12 2021] 616186/616186 (100%) pairs aligned
[Wed May 12 13:08:12 2021] Alignment of pairs finished
[Wed May 12 13:08:12 2021] Translating coordinates from local pair level to contig level
[Wed May 12 13:08:19 2021] Finished spliced alignment
[Wed May 12 13:08:19 2021] Flagging top chains
error: Gene-protein pair "37843_g-sp_A2EF582_PFP3_TRIVA_Pyrophosphate--fructose_6-phosphate_1-phosphotransferase_3__AltName:_Full=6-phosphofructokinase,_pyrophosphate_dependent_3__AltName:_Full=PPi-dependent_phosphofructokinase_3__Short=PPi-PFK_3__AltName:_Full=Pyrophosphate-dependent_6-" present in the Spaln output was not found in the file with DIAMOND gene-protein pairs. This issue can be caused by the presence of special characters in the fasta headers of input files. Please remove any special characters and re-run ProtHint. See https://github.com/gatech-genemark/ProtHint#input for more details about the input format.
[Wed May 12 13:08:21 2021] error: ProtHint exited due to an error in command: /opt/ProtHint-2.6.0/bin/flag_top_proteins.py Spaln/spaln.gff /scratch/cgsb/carlton/steven/Tv_PacBioPGAJelly/final_polished_assembly/braker2_May5_pipelineD_1/diamond/diamond.out > tmp
ERROR in file /opt/BRAKER-2.1.6/scripts/braker.pl at line 5346
Failed to execute /opt/ProtHint-2.6.0/bin/prothint.py --threads=10 --geneMarkGtf /scratch/cgsb/carlton/steven/Tv_PacBioPGAJelly/final_polished_assembly/braker2_May5_pipelineD_1/GeneMark-ES/genemark.gtf /scratch/cgsb/carlton/steven/Tv_PacBioPGAJelly/final_polished_assembly/braker2_May5_pipelineD_1/genome.fa /scratch/cgsb/carlton/steven/Tv_PacBioPGAJelly/final_polished_assembly/braker2_May5_pipelineD_1/proteins.fa!
It appears that some process (ProtHint?) does not like a FASTA header of one protein in my protein set. I will shorten that header (and clean any other messy headers) and try again.
FYI, the editing worked and in the end this BRAKER pipeline D run did complete, but it generated an implausible number of genes (>75,000, a third of which were located in masked regions) and many implausible models (most models had 0-2 introns, which accords with older data, but many also had >20 introns, as many as 62 -- and these genes were often located in nonmasked regions)
Using a much smaller protein evidence set -- eliminating the OrthoDB protein records, leaving only 375 manually curated same-species records -- brought the output gene number down by ~half, but the maximum #CDS/gene remained high, >40
I am wondering if pipeline D is appropriate for this organism.
Did you have a chance to run BUSCO or any other quality assessment tool for both outputs? I wonder whether the noisy output from pipeline D also contains more BUSCO genes than the one with close proteins only.
There is also a new way of combining protein and RNA inputs, with TSEBRA: https://www.biorxiv.org/content/10.1101/2021.06.07.447316v1. That should lead to better results than pipeline D.
No, I did not; I have since abandoned using RNA and protein inputs together. I can't use TSEBRA because running BRAKER in RNA evidence only mode (BRAKER1) fails at the GeneMark step (as Dr. Hoff predicted to me), since the only use BRAKER makes of RNA-Seq mapping data is for predicting introns. My genome is extremely intron-poor, causing GeneMark to fail. I have not figured out how to alter extrinsic.cfg to employ RNA-seq mapping data for other uses, eg transcript prediction (similar to what Stringtie does). Meanwhile, running BRAKER in protein-evidence-only mode (BRAKER1) either fails or succeeds depending on the size and contents of the evidence file. Using the Protozoa subset of OrthoDB as evidence (>1.7 million fasta records) produces an even more enormous number (>95,000) of gene models, though on the positive side, the maximum #CDS/gene went down to 2. Using a far smaller set of closely-related proteins (553 fasta records) caused GeneMark to fail due to lack of introns. (error message: #WARNING: #The hints file(s) for GeneMark-EX contain less than 150 introns with multiplicity >= 4! (In total, 88 unique introns are contained. 7 have a multiplicity >= 4.) )
My conclusion is that BRAKER isn't easily used with eukaryotic genomes that have few introns.
You can run BRAKER without any evidence to solve the problem that genemark fails. After the run has finished (ignore the output, you only want to have the resulting species parameter set) take this hints file from the failed braker run and the species set from the success run and use both to run predictions only with braker. Do it once for RNAseq, once for proteins, then use TSEBRA (but not with the default parameter set, there is a more relaxed parameter set for species with less evidence).
On Mon 21. Jun 2021 at 23:17, krabapple @.***> wrote:
No, I did not; I have since abandoned using RNA and protein inputs together. I can't use TSEBRA because running BRAKER in RNA evidence only mode (BRAKER1) fails at the GeneMark step (as Dr. Hoff predicted to me), since the only use BRAKER makes of RNA-Seq mapping data is for predicting introns. My genome is extremely intron-poor, causing GeneMark to fail. I have not figured out how to alter extrinsic.cfg to employ RNA-seq mapping data for other uses, eg transcript prediction (similar to what Stringtie does). Meanwhile, running BRAKER in protein-evidence-only mode (BRAKER1) either fails or succeeds depending on the size and contents of the evidence file. Using the Protozoa subset of OrthoDB as evidence (>1.7 million fasta records) produces an enourmous number (>95,000) of gene models, though on the positive side, the maximum #CDS/gene went down to 2. Using a far smaller set of closely-related proteins (553 fasta records) caused GeneMark to fail due to lack of introns. [error message: "The hints file(s) for GeneMark-EX contain less than 1000 introns. (In total, 88 unique introns are contained.) Genemark-EX might fail due to the low number of hints."]
— You are receiving this because you are subscribed to this thread. Reply to this email directly, view it on GitHub https://github.com/Gaius-Augustus/BRAKER/issues/357#issuecomment-865350255, or unsubscribe https://github.com/notifications/unsubscribe-auth/AJMC6JEI4YYTYZLMHVIBF7TTT6T7FANCNFSM43I6VEBQ .
Hello, thank you for the suggestion. To help me attempt it, can you clarify the steps? I am confused as to what you mean by the 'success run' and the 'failed run' in this case.
The ab initio run you are suggesting I do, is that the 'success run'? (and I should use the species folder/file generated by this?) And my failed RNAevidence-only BRAKER1 run, is that the 'failed run' ? (and I should use the hints generated by this?)
If not, what runs are you referring to?