ReducedExperiment
Update the following URL to point to the GitHub repository of the package you wish to submit to Bioconductor
- Repository: https://github.com/jackgisby/ReducedExperiment
Confirm the following by editing each check box to '[x]'
-
[x] I understand that by submitting my package to Bioconductor, the package source and all review commentary are visible to the general public.
-
[x] I have read the Bioconductor Package Submission instructions. My package is consistent with the Bioconductor Package Guidelines.
-
[x] I understand Bioconductor Package Naming Policy and acknowledge Bioconductor may retain use of package name.
-
[x] I understand that a minimum requirement for package acceptance is to pass R CMD check and R CMD BiocCheck with no ERROR or WARNINGS. Passing these checks does not result in automatic acceptance. The package will then undergo a formal review and recommendations for acceptance regarding other Bioconductor standards will be addressed.
-
[x] My package addresses statistical or bioinformatic issues related to the analysis and comprehension of high throughput genomic data.
-
[x] I am committed to the long-term maintenance of my package. This includes monitoring the support site for issues that users may have, subscribing to the bioc-devel mailing list to stay aware of developments in the Bioconductor community, responding promptly to requests for updates from the Core team in response to changes in R or underlying software.
-
[x] I am familiar with the Bioconductor code of conduct and agree to abide by it.
I am familiar with the essential aspects of Bioconductor software management, including:
- [x] The 'devel' branch for new packages and features.
- [x] The stable 'release' branch, made available every six months, for bug fixes.
- [x] Bioconductor version control using Git (optionally via GitHub).
For questions/help about the submission process, including questions about the output of the automatic reports generated by the SPB (Single Package Builder), please use the #package-submission channel of our Community Slack. Follow the link on the home page of the Bioconductor website to sign up.
Hi @jackgisby
Thanks for submitting your package. We are taking a quick look at it and you will hear back from us soon.
The DESCRIPTION file for this package is:
Package: ReducedExperiment
Type: Package
Title: Containers and tools for dimensionally-reduced -omics data
Version: 0.99.0
Authors@R: c(
person("Jack", "Gisby", , "[email protected]", role = c("aut", "cre"),
comment = c(ORCID = "0000-0003-0511-8123")),
person("Michael", "Barnes", , "[email protected]", role = c("aut"),
comment = c(ORCID = "0000-0001-9097-7381"))
)
Date: 2024-07-30
Description: Provides SummarizedExperiment-like containers for storing and
manipulating dimensionally-reduced assay data. The ReducedExperiment
classes allow users to simultaneously manipulate their original dataset
and their decomposed data, in addition to other method-specific outputs
like feature loadings. Implements utilities and specialised classes for the
application of stabilised independent component analysis (sICA) and
weighted gene correlation network analysis (WGCNA).
License: GPL (>= 3)
Encoding: UTF-8
Depends:
R (>= 4.4.0),
SummarizedExperiment,
methods
Imports:
WGCNA,
ica,
moments,
clusterProfiler,
msigdbr,
RColorBrewer,
car,
lme4,
lmerTest,
pheatmap,
biomaRt,
stats,
grDevices,
BiocParallel,
ggplot2,
patchwork,
BiocGenerics,
S4Vectors
Suggests:
knitr,
rmarkdown,
testthat,
biocViews,
BiocCheck,
BiocStyle,
airway
Roxygen: list(markdown = TRUE)
RoxygenNote: 7.3.1
VignetteBuilder: knitr
URL: https://github.com/jackgisby/ReducedExperiment
BugReports: https://github.com/jackgisby/ReducedExperiment/issues
biocViews: GeneExpression, Infrastructure, DataRepresentation, Software,
DimensionReduction, Network
Your package has been added to git.bioconductor.org to continue the pre-review process. A build report will be posted shortly. Please fix any ERROR and WARNING in the build report before a reviewer is assigned or provide a justification on why you feel the ERROR or WARNING should be granted an exception.
IMPORTANT: Please read this documentation for setting up remotes to push to git.bioconductor.org. All changes should be pushed to git.bioconductor.org moving forward. It is required to push a version bump to git.bioconductor.org to trigger a new build report.
Bioconductor utilized your github ssh-keys for git.bioconductor.org access. To manage keys and future access you may want to active your Bioconductor Git Credentials Account
Dear Package contributor,
This is the automated single package builder at bioconductor.org.
Your package has been built on the Bioconductor Single Package Builder.
On one or more platforms, the build results were: "WARNINGS". This may mean there is a problem with the package that you need to fix. Or it may mean that there is a problem with the build system itself.
Please see the build report for more details.
The following are build products from R CMD build on the Single Package Builder: Linux (Ubuntu 24.04.1 LTS): ReducedExperiment_0.99.0.tar.gz
Links above active for 21 days.
Remember: if you submitted your package after July 7th, 2020,
when making changes to your repository push to
[email protected]:packages/ReducedExperiment to trigger a new build.
A quick tutorial for setting up remotes and pushing to upstream can be found here.
A reviewer has been assigned to your package for an indepth review. Please respond accordingly to any further comments from the reviewer.
Received a valid push on git.bioconductor.org; starting a build for commit id: 10c45474a78fa8d0dda4a4fe084b9abe1c18f931
Received a valid push on git.bioconductor.org; starting a build for commit id: aaca6e50909ef5814f556f72ce09be19c82f5111
Dear Package contributor,
This is the automated single package builder at bioconductor.org.
Your package has been built on the Bioconductor Single Package Builder.
Congratulations! The package built without errors or warnings on all platforms.
Please see the build report for more details.
The following are build products from R CMD build on the Single Package Builder: Linux (Ubuntu 24.04.1 LTS): ReducedExperiment_0.99.1.tar.gz
Links above active for 21 days.
Remember: if you submitted your package after July 7th, 2020,
when making changes to your repository push to
[email protected]:packages/ReducedExperiment to trigger a new build.
A quick tutorial for setting up remotes and pushing to upstream can be found here.
Dear Package contributor,
This is the automated single package builder at bioconductor.org.
Your package has been built on the Bioconductor Single Package Builder.
On one or more platforms, the build results were: "WARNINGS". This may mean there is a problem with the package that you need to fix. Or it may mean that there is a problem with the build system itself.
Please see the build report for more details.
The following are build products from R CMD build on the Single Package Builder: Linux (Ubuntu 24.04.1 LTS): ReducedExperiment_0.99.2.tar.gz
Links above active for 21 days.
Remember: if you submitted your package after July 7th, 2020,
when making changes to your repository push to
[email protected]:packages/ReducedExperiment to trigger a new build.
A quick tutorial for setting up remotes and pushing to upstream can be found here.
Received a valid push on git.bioconductor.org; starting a build for commit id: bc7c6edb3e99c028b8a8e581fbdb9d30387bf250
Dear Package contributor,
This is the automated single package builder at bioconductor.org.
Your package has been built on the Bioconductor Single Package Builder.
Congratulations! The package built without errors or warnings on all platforms.
Please see the build report for more details.
The following are build products from R CMD build on the Single Package Builder: Linux (Ubuntu 24.04.1 LTS): ReducedExperiment_0.99.3.tar.gz
Links above active for 21 days.
Remember: if you submitted your package after July 7th, 2020,
when making changes to your repository push to
[email protected]:packages/ReducedExperiment to trigger a new build.
A quick tutorial for setting up remotes and pushing to upstream can be found here.
I have pushed updates to reduce the time taken to run checks. In addition, I thought it might be worth explaining the notes produced during the checks. I provide reasoning for not further addressing these, but I am of course happy to make further changes.
R CMD Check
- "Unexported objects imported by ':::' calls" - These are included because we extend the SummarizedExperiment object slice methods. In the original SummarizedExperiment they use these functions in the method (see BiocGenerics:::replaceSlots, SummarizedExperiment:::.SummarizedExperiment.charbound). I included these functions using ::: because I felt it made more sense to stick as close as possible to the original SummarizedExperiment method. I could reimplement the .SummarizedExperiment.charbound function, but there is then risk that the original function is changed and we don't modify this package's version to reflect the change - also this function itself calls an internal S4Vectors function.
BiocCheck
- "Avoid 1:...; use seq_len() or seq_along()" - I use seq_len/seq_along in the code. However, this specific instance is the default value for a function argument. For the default, there is no difference between seq_len(30) and 1:30, and if the user changes this default it is up to them to pass a valid argument. Note that this argument is passed directly to WGCNA::pickSoftThreshold.
- "Use accessors; don't access S4 class slots via '@' in examples/vignettes." - This note is referring to the part of the vignette where we demonstrate how to create an object that inherits from ReducedExperiment, and so we demonstrate how a user could make their own accessor method.
- "The recommended function length is 50 lines or less. There are 12 functions greater than 50 lines." - Most of these functions are only marginally above the limit. In some cases they could be made shorter, but I felt that this would be at the cost of comments and/or readability. In some cases I split functions into subfunctions, but for the remaining I felt that doing so would increase complexity. The worst offenders are the slice and slice replacement methods (e.g., 'setReplaceMethod("["...'). I wrote these to be consistent with the parent SummarizedExperiment methods, although I did encapsulate some repeat logic in a separate function to increase readability and reduce the function lengths.
- "Consider adding these automatically suggested biocViews: Sequencing, Coverage" - We do not directly use sequencing data, so I instead used the "GeneExpression" biocView. I do not believe the "Coverage" tag is relevant.
- "Consider shorter lines; 43 lines (1%) are > 80 characters long." - I have enforced this rule for the code, this note refers to the roxygen strings. The roxygen strings are still reasonable (<100 chars) but not always under this limit.
- "Update R version dependency from 4.4.0 to 4.5.0." - This would make it challenging for people to install the package via GitHub until it is released on Bioconductor.
Other
- In the vignette we have four eval=FALSE chunks. Two of these are for installing the package. One is for running the SummarizedExperiment vignette (we recommend users to read it first). The last is for the estimate_stability method. This is a very computationally intensive function. We run it for a very small dataset as a test, and we do the same for the function's example code. In the vignette we wanted to demonstrate how to interpret the output of this function. But the output is only really interpretable for "real" datasets with a minimal number of samples / iterations. For this reason, we don't run the code in the vignette. We instead show the result of running the same code on the full dataset. In theory I could change this so that we both run the function in the vignette for a very small number of iterations, and then show the actual interpretable plot afterwards. But I felt this would be confusing.
Thank you, @jackgisby, for your explanations. I haven't yet started my review, but I will have it for you within 3 weeks (usually sooner).
Cheers, Pete
Hi @jackgisby,
Thank you for submitting ReducedExperiment to Bioconductor.
I've completed my checklist review of ReducedExperiment below and I have separated the issues into Required and Recommended points that I would ask you to address before the package can be accepted. Technically, the package is in great shape - you've clearly taken the effort to follow best practises and developed it to a high standard - so I don't foresee any troubles for you to address these points.
The main issue I'd like to discuss with you is where ReducedExperiment fits in with existing Bioconductor packages and whether it's necessary that it re-implements what I see as perhaps overlapping functionality with existing packages. Please do consider this a discussion - I'm not asking you to make immediate, wholesale changes - but I'm providing a perspective from a potential user that I hope is helpful to you as a developer.
Would you please provide line-by-line comments to my initial review so that I know what changes I'm looking for in my re-review.
Cheers, Pete
Relationship of ReducedExperiment to existing Bioconductor packages and functionality
My brief summary of ReducedExperiment is that it:
- Introduces the ReducedExperiment class, an extension of SummarizedExperiment, and 2 derived subclasses (FactorisedExperiment and ModularExperiment) for storing dimensionality reduction results of omics data.
- Provides wrappers of ica and WCGNA functionality that use these classes.
Now, the SingleCellExperiment class, in the package of the same name (and part of Bioconductor for 7 years), is an extension of the SummarizedExperiment class that includes a reducedDims slot; see help("reducedDims", package = "SingleCellExperiment") for details.
SingleCellExperiment implements this concept in a different way to ReducedExperiment - most obviously in that it doesn't use a third dimension to represent the number of factors - but conceptually it feels very similar to me.
Admittedly, the name SingleCellExperiment sure sounds as though it precludes non-single cell data, but it's just a name and there's no technical reason not to use it for, say, bulk RNA-seq data (I do myself for some analysis in my day job).
So my main questions are:
- Did you consider using SingleCellExperiment object instead of creating new classes?
- If so, would you please summarise why you felt it didn't suit your needs?
At the very least, the vignette needs a brief review of and comparison to SingleCellExperiment and the reducedDims slot of the SingleCellExperiment class.
Finally, and perhaps less directly relevant, as a developer, the BiocSingular and its LowRankMatrix class may be of interest to you. Noting that not all the the dimensionality reductions considered by ReducedExperiment may be suitable/compatible for/with the SVD/PCA-focused BiocSingular.
Required
- [ ] The 'Installation' section of the vignette should focus on just the installation (giving the 'official'
BiocManager::install()installation instructions priority over thedevtools::install_github()-based instructions), not package loading or setting preferences for ggplot2. - [ ] When I worked through the vignette, the output of first figure didn't match the inserted image from https://raw.githubusercontent.com/jackgisby/ReducedExperiment/devel/inst/stability.png, presumably because different
BPPARAMwas used when generating the hosted image (particularly theRNGseedcomponent)? - [ ] Functions should not set a default seed. E.g.,
estimate_stability()has default parameter ofBPPARAM = BiocParallel::SerialParam(RNGseed = 1)but should just useBPPARAM = BiocParallel::SerialParam(). - [ ] Pre-computed results should use local assets rather than web-hosted assets. E.g., something like
knitr::include_graphics(system.file("stability.png", package = "ReducedExperiment"))instead of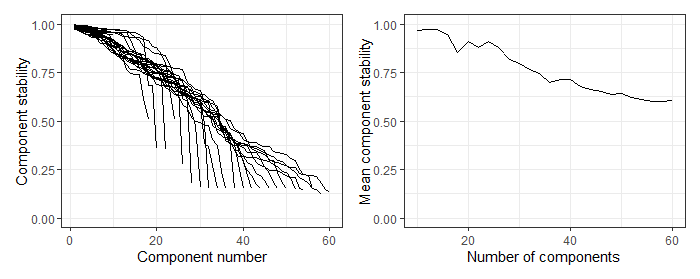; see https://r-pkgs.org/vignettes.html#filepaths. - [ ] What's
inst/dockerneeded for? - [ ] Please remove the
dir.create("tempOutput")in the vignette. It doesn't seem to be used elsewhere in the vignette. Moreover,tempdir()should be used for any temporary outputs that are required.
Recommended
- [ ] The
@inheritParamsroxygen2 tag may be helpful for documenting the wrapper functions (e.g., documenting thepowerVectorargument ofassess_soft_threshold()); see https://roxygen2.r-lib.org/articles/reuse.html?q=inheritparam#inheriting-from-other-packages. - [ ] Why are function names are a mixture of
snake_case()andcamelCase()? It's generally best within a package to pick one format and stick with it. - [ ] Strongly recommend adding figure captions to the vignette (via the
fig.capchunk option). - [ ] The vignette plots of
associations_feandassociations_meuse adjusted P-value but the figure's axis labels refer to 'P-value' rather than 'adjusted P-value'. Recommend fixing the axis labels. - [ ] Strongly recommend checking the formatting of rendered vignette to make sure it looks as you intend (e.g., in Section 4 of the vignette I see what appears to be raw markdown that is supposed to be formatted as a list).
- [ ] Consider adding a package man page; see https://contributions.bioconductor.org/docs.html#package-level-documentation
- Nothing to do for this one, but
covr::report()may be useful to identify untested parts of the package.
Hi @jackgisby,
I am on leave for the Christmas / New Year break where I work, so please don't expect a reply from me until after I return to work on January 2.
Cheers, Pete
Thanks very much for your detailed review @PeteHaitch. I am taking time to consider your points, but I am also on leave until January 6, so it might take some time for me to complete my response.
Received a valid push on git.bioconductor.org; starting a build for commit id: 7519a2802265b185c3f16f802c7c77170747e68e
Dear Package contributor,
This is the automated single package builder at bioconductor.org.
Your package has been built on the Bioconductor Single Package Builder.
Congratulations! The package built without errors or warnings on all platforms.
Please see the build report for more details.
The following are build products from R CMD build on the Single Package Builder: Linux (Ubuntu 24.04.1 LTS): ReducedExperiment_0.99.4.tar.gz
Links above active for 21 days.
Remember: if you submitted your package after July 7th, 2020,
when making changes to your repository push to
[email protected]:packages/ReducedExperiment to trigger a new build.
A quick tutorial for setting up remotes and pushing to upstream can be found here.
Hi @PeteHaitch,
Thanks for your detailed review.
I have made changes in response to the required and recommended changes, as detailed below. I also carefully considered your point about overlapping functionality with existing frameworks. I have provided a detailed explanation of my rationale for creating the ReducedExperiment methods based on the SummarizedExperiment system, rather than simply using existing functionality in packages like SingleCellExperiment.
Choice of underlying framework
I certainly agree that there is overlapping functionality between ReducedExperiment and single-cell frameworks, particularly in their ability to store low-dimensional representations (LDRs) of data. My original goal with the package was to create a system that provided a convenient framework for applying interpretable dimensionality reduction approaches and manipulating their outputs. I considered multiple systems, including SingleCellExperiment, to base these approaches on. I decided that I required method-specific functionality that I felt would have been difficult to implement within existing frameworks. Primarily, they place weak assumptions on the nature of the stored LDRs and generally only have the capacity to store sample-level scores.
While there are similarities in implementation, packages like SingleCellExperiment approach dimensionality reduction quite differently. In single-cell analysis, LDRs primarily serve to reduce noise, enable visualization, and facilitate downstream analysis like clustering. The actual interpretation of the low-dimensional components is typically not the focus. In contrast, ReducedExperiment was specifically designed for methods like ICA and WGCNA where interpreting the components themselves is the primary goal.
This philosophical difference is reflected in the implementations: • A SingleCellExperiment (SCE) allows multiple LDRs with minimal assumptions about their generation method. • A ReducedExperiment contains a single LDR with strong assumptions about its structure and generation method, and the capacity to store additional method-specific information pertaining to this LDR.
I considered the following questions:
- “Could I have created the methods and workflows using an existing class system?”
- “Could I have minimised overlapping functionality by basing the ReducedExperiment classes on another framework, like SCE?”
In the first scenario, I could have implemented workflows for interpretable LDR analysis storing sample-level scores in an SCE. The SCE package implements the reduced.dim.matrix object, which allows attributes of the sample-level scores to be retained when the object is sliced. However, these attributes themselves are not sliced accordingly and will become misaligned with these scores and the SCE assay object. I believe it would have been difficult to ensure that this metadata remained valid when being manipulated by users and ensure matching of the correct method to the type of LDR. Overall, I think these workflows would have been more difficult to implement, less convenient to use and more error-prone if based on SCE.
I think the second scenario would have been more viable but would have been difficult to implement robustly. Among these issues:
- Many of the methods implemented in the ReducedExperiment workflows rely on method-specific slots. This would be challenging to implement in a data structure that allows for multiple LDRs, each generated by their own method with different accompanying data structures.
- Frameworks like SCE implement additional features that would have added complexity for our use case. For instance, SCE allows alternative feature sets that can be swapped with the active features. Many ReducedExperiment methods rely on exact correspondence between features and reduced dimensions (e.g., in the loadings/rotation matrix), which is verified through validity checks. Supporting this functionality while maintaining compatibility with SCE's alternative feature sets would have added considerable complexity to the implementation.
- As an example, by default the FactorisedExperiment class implements enrichment methods specifically designed for ICA components using feature loadings, while ModularExperiment implements overrepresentation tests based on the module assignments slot. These method-specific analyses rely on: 1) slots not implemented in SCE (e.g., loadings); 2) a way to match the methods with the type of LDR; 3) maintenance of correspondence between features and reduced dimensions.
- While a minor point, the third dimension in ReducedExperiment enables coordinated manipulation of related data. Our implementation allows the objects to be sliced by components, whereas this is not the case in an SCE. For instance, when filtering components in a FactorisedExperiment, we automatically filter the corresponding loadings and stability values, and when subsetting features in a ModularExperiment, we filter the feature-module assignments. This would be challenging to achieve in an SCE that contains multiple distinct LDRs each with their own method-specific slots.
- I think there are some other minor benefits to basing the package on SummarizedExperiment, such as avoiding extraneous functionality that would not be relevant to the typical user of ReducedExperiment (e.g., cell-cell pairings) and making it clearer to users that the package is appropriate for bulk data.
Possible approaches to extending SCE for our purposes would have included:
- Basing the ReducedExperiment classes on SCE, while limiting them to a single LDR and disabling certain SCE properties. While technically viable, it would likely increase the complexity of the ReducedExperiment implementation, as opposed to reducing it by making use of shared functionality. The resulting objects would probably no longer interact well with other packages that work with SCEs.
- Using SCE as a base, but create new objects for each LDR method that reside in its reduced dimensions slot. These objects could implement their respective methods and validity checks. I think this is a more interesting idea, but I believe it would have been challenging to coordinate changes to the upstream SCE object, such as slicing of features or swapping of feature sets, and maintain validity.
Perhaps part of the issue is that the package headline sells itself primarily as an object system, when really the aim of the package is to provide a framework for workflows for interpretable dimensionality reduction. I think it’s worth noting that we don’t just provide wrappers around ICA - the package includes implementations of stabilised ICA (sICA) and the Maximally Stable Transcriptome Dimension (MSTD) approach and associated analyses and visualisations, which were previously only available in Python and MATLAB. The object structure was designed to support these and similar analytical approaches while maintaining strict validity checks on the relationships between features, samples, and components. I could make changes to the package descriptions that might make the package’s purpose/benefits clearer and more distinct?
In summary, while SCE has overlapping functionality, I felt that a system that placed stricter assumptions on the content of the LDRs to facilitate interpretable dimensionality reduction was necessary. I believe the package provides useful novel functionality (e.g., sICA/MSTD) and that it integrates well with SummarizedExperiment. I also think that, given the differences in how SCE and ReducedExperiment treat LDRs, it would be challenging to adapt the workflows at this point to be based on SCE objects. I have added a section to the vignette comparing ReducedExperiment with existing frameworks to help users understand the differences and when to use each framework.
Additionally, thanks for the BiocSingular suggestion. I think this could be useful if we extend FactorisedExperiment to include PCA, or find a way to adapt it to ICA.
Required changes
- [x] The 'Installation' section of the vignette should focus on just the installation (giving the 'official' BiocManager::install() installation instructions priority over the devtools::install_github()-based instructions), not package loading or setting preferences for ggplot2.
I have reorganized the vignette as instructed. I also made a minor change to the README to prioritise the Bioconductor installation routes.
- [x] When I worked through the vignette, the output of first figure didn't match the inserted image from https://raw.githubusercontent.com/jackgisby/ReducedExperiment/devel/inst/stability.png, presumably because different BPPARAM was used when generating the hosted image (particularly the RNGseed component)?
Fixed, users should now be able to replicate it with the vignette code. I have changed the code to explicitly specify the RNGseed. I made slight changes to the surrounding text to reflect the changes.
- [x] Functions should not set a default seed. E.g., estimate_stability() has default parameter of BPPARAM = BiocParallel::SerialParam(RNGseed = 1) but should just use BPPARAM = BiocParallel::SerialParam().
I have removed the RNGseed argument from the function parameters of estimate_stability and run_ica. I have added these to the tests/vignettes instead and made it clear in the documentation that this needs to be specified for reproducibility.
- [x] Pre-computed results should use local assets rather than web-hosted assets. E.g., something like knitr::include_graphics(system.file("stability.png", package = "ReducedExperiment")).
This has now been fixed.
- [x] What's inst/docker needed for?
I set up GitHub Actions to automatically build a docker image for the development version of the package, based on the Bioconductor image, and push it to Docker Hub. I use this for integrating the package with Nextflow pipelines. The directory structure was based on that in other packages (e.g., github.com/sbg/sevenbridges-r). I have now modified the Dockerfile to add metadata in the same style as the parent image and I have modified the Actions workflow to append the package version to the docker images. This should make the images useful to others.
- [x] Please remove the dir.create("tempOutput") in the vignette. It doesn't seem to be used elsewhere in the vignette. Moreover, tempdir() should be used for any temporary outputs that are required.
Removed.
Recommended changes
- [x] The @inheritParams roxygen2 tag may be helpful for documenting the wrapper functions (e.g., documenting the powerVector argument of assess_soft_threshold()); see https://roxygen2.r-lib.org/articles/reuse.html?q=inheritparam#inheriting-from-other-packages.
I have now applied @inheritParams to both incorporate documentation for wrapper functions and reduce redundancy for within-package documentation.
- [x] Why are function names are a mixture of snake_case() and camelCase()? It's generally best within a package to pick one format and stick with it.
I have now switched everything to camelCase.
- [x] Strongly recommend adding figure captions to the vignette (via the fig.cap chunk option).
Added.
- [x] The vignette plots of associations_fe and associations_me use adjusted P-value but the figure's axis labels refer to 'P-value' rather than 'adjusted P-value'. Recommend fixing the axis labels.
Fixed. The labels now specify adjusted P-value.
- [x] Strongly recommend checking the formatting of rendered vignette to make sure it looks as you intend (e.g., in Section 4 of the vignette I see what appears to be raw markdown that is supposed to be formatted as a list).
Fixed. The bullet points now appear correctly.
- [ ] Consider adding a package man page; see https://contributions.bioconductor.org/docs.html#package-level-documentation
In theory I am happy to do this but a package man page would conflict with the class documentation. Similarly to SummarizedExperiment, my main class (ReducedExperiment) has the same name as the package. Therefore, when you type ?ReducedExperiment, it gives you the documentation for the class. I think this might be a reasonable entry point for the package, so this might be better left as-is.
- [x] Nothing to do for this one, but covr::report() may be useful to identify untested parts of the package.
I set up GitHub Actions to automatically run tests and assess coverage, which can be seen here: https://app.codecov.io/gh/jackgisby/ReducedExperiment/tree/devel/R I think the coverage is sufficient for now (>90%) but in future I will consider targeting untested code.
Thank you for your considered and detailed reply, @jackgisby. I'll reply properly in the next few days to continue the review.
Hi @jackgisby,
Thank you making the required changes, considering the recommended changes, and for including a summary of where ReducedExperiment fits in with existing Bioconductor packages in the vignette.
I'm now happy to accept ReducedExperiment into Bioconductor. Congratulations and thank you for your contribution!
A few minor things to consider:
- [ ] In the vignette, should
set.seed(1)be in ainclude=TRUEchunk if it is required (but perhaps it is no longer required)? - [ ] Similarly, the
load-packageschunk should ainclude=TRUEchunk (otherwise a user copy+pasting the vignette code will get an error, e.g., when they try to runggplot()) - [ ] Why does
wave1_fit_indices <- assessSoftThreshold(wave1_se, verbose = 0)still print intermediate output? How can a user completely supress this output viaverbose?
Cheers, Pete
Your package has been accepted. It will be added to the Bioconductor nightly builds.
Thank you for contributing to Bioconductor!
Reviewers for Bioconductor packages are volunteers from the Bioconductor community. If you are interested in becoming a Bioconductor package reviewer, please see Reviewers Expectations.
cannot build unless issue is open and has the 'pre-review' label or '2. review in progress' label, or is closed and has the 'TESTING' label.
you can ignore this message it just means the package was accepted and changes were made after the fact that the reviewer might want to glance at. Once accepted there are no on demand builds and it will be moved onto the official daily builder to become part of the available Bioconductor packages
Hi @PeteHaitch,
Great - thanks for your help and suggestions. I made some more changes in response to your points.
- [x] In the vignette, should
set.seed(1)be in ainclude=TRUEchunk if it is required (but perhaps it is no longer required)?
I have now removed this.
- [x] Similarly, the
load-packageschunk should ainclude=TRUEchunk (otherwise a user copy+pasting the vignette code will get an error, e.g., when they try to runggplot())
This is now included.
- [x] Why does
wave1_fit_indices <- assessSoftThreshold(wave1_se, verbose = 0)still print intermediate output? How can a user completely suppress this output viaverbose?
The underlying WGCNA function had a print statement not conditional on verbose. I have modified the function to use capture.output so that, when verbose is 0 (default), the print statement is captured.
The default branch of your GitHub repository has been added to Bioconductor's git repository as branch devel.
To use the git.bioconductor.org repository, we need an 'ssh' key to associate with your github user name. If your GitHub account already has ssh public keys (https://github.com/jackgisby.keys is not empty), then no further steps are required. Otherwise, do the following:
See further instructions at
https://bioconductor.org/developers/how-to/git/
for working with this repository. See especially
https://bioconductor.org/developers/how-to/git/new-package-workflow/ https://bioconductor.org/developers/how-to/git/sync-existing-repositories/
to keep your GitHub and Bioconductor repositories in sync.
Your package will be included in the next nigthly 'devel' build (check-out from git at about 6 pm Eastern; build completion around 2pm Eastern the next day) at
https://bioconductor.org/checkResults/
(Builds sometimes fail, so ensure that the date stamps on the main landing page are consistent with the addition of your package). Once the package builds successfully, you package will be available for download in the 'Devel' version of Bioconductor using BiocManager::install("ReducedExperiment"). The package 'landing page' will be created at
https://bioconductor.org/packages/ReducedExperiment
If you have any questions, please contact the bioc-devel mailing list (https://stat.ethz.ch/mailman/listinfo/bioc-devel); this issue will not be monitored further.