velocyto.py
 velocyto.py copied to clipboard
velocyto.py copied to clipboard
index error list out of range
Hi this is a similar to issue to some of the GTF error issues other people have raised but I saw the solution was removing a field from the annotation file, however I don't think I have that field so I am confused what I should do.
This is my error below, and I am attaching a screenshot of my annotation file:
velo.sh velo.sh.e4851593 velo.sh.o4851593
-bash-4.2$ cat velo.sh.e4851593
Traceback (most recent call last):
File "/nfs/sw/velocyto/velocyto-0.17/bin/velocyto", line 10, in 
I also have this error with 10X data:
File "/usr/local/bin/velocyto", line 11, in
Hi @kaizen89 and @rekham1077, I think we might be discussing a similar error on the Open Issue #135. Check it out and see if it is helpful. :]
Hi @kaizen89 I actually ended up getting it to run, but I had to re-do my alignment with a different annotation file. I had originally done it with the annotation from UCSC but I re-aligned with cellranger v3 reference and annotation so those should work you.
But I am using a different version of velocyto, 0.17.15.
Hello,
@rekham1077. I am also facing the same issue when running: velocyto run-smartseq2:
Traceback (most recent call last): File "/home/jihoonk/miniconda3/envs/py37/bin/velocyto", line 8, in <module> sys.exit(cli()) File "/home/jihoonk/miniconda3/envs/py37/lib/python3.7/site-packages/click/core.py", line 829, in __call__ return self.main(*args, **kwargs) File "/home/jihoonk/miniconda3/envs/py37/lib/python3.7/site-packages/click/core.py", line 782, in main rv = self.invoke(ctx) File "/home/jihoonk/miniconda3/envs/py37/lib/python3.7/site-packages/click/core.py", line 1259, in invoke return _process_result(sub_ctx.command.invoke(sub_ctx)) File "/home/jihoonk/miniconda3/envs/py37/lib/python3.7/site-packages/click/core.py", line 1066, in invoke return ctx.invoke(self.callback, **ctx.params) File "/home/jihoonk/miniconda3/envs/py37/lib/python3.7/site-packages/click/core.py", line 610, in invoke return callback(*args, **kwargs) File "/home/jihoonk/miniconda3/envs/py37/lib/python3.7/site-packages/velocyto/commands/run_smartseq2.py", line 74, in run_smartseq2 samtools_memory=1, dump=dump, loom_numeric_dtype=dtype, verbose=verbose, additional_ca=additional_ca) File "/home/jihoonk/miniconda3/envs/py37/lib/python3.7/site-packages/velocyto/commands/_run.py", line 229, in _run results = exincounter.count(bamfile_cellsorted, multimap=multimap) # NOTE: we would avoid some millions of if statements evaluations if we write two function count and count_with output File "/home/jihoonk/miniconda3/envs/py37/lib/python3.7/site-packages/velocyto/counter.py", line 761, in count dict_layer_columns, list_bcs = self.count_cell_batch() File "/home/jihoonk/miniconda3/envs/py37/lib/python3.7/site-packages/velocyto/counter.py", line 1167, in _count_cell_batch_non_stranded self.logic.count(molitem, bcidx, dict_layers_columns, self.geneid2ix) File "/home/jihoonk/miniconda3/envs/py37/lib/python3.7/site-packages/velocyto/logic.py", line 1091, in count upstream_exon = segment_match.feature.get_upstream_exon() File "/home/jihoonk/miniconda3/envs/py37/lib/python3.7/site-packages/velocyto/feature.py", line 75, in get_upstream_exon return self.transcript_model.list_features[ix] IndexError: list index out of range
I am using the mm10.refflat.gtf that was used for STAR alignment (to generate the bam files per cell), which looks like:
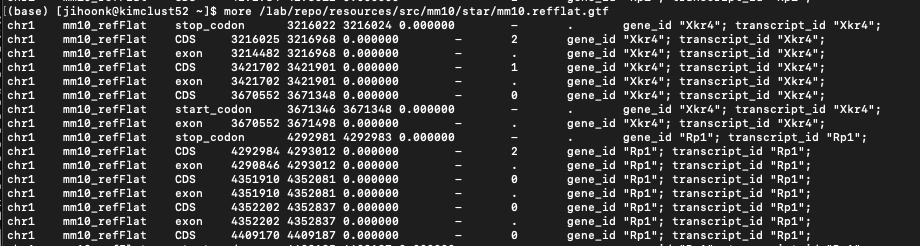
To my knowledge, the gtf follows the requirements for velocyto (described here). I'm not sure what I should do.
Hi @RBBurl1227 and @rekham1077 -- When you said you redo the alignment with new annotation file, did you mean you rerun the cellranger count or you meant you just use a different genes/genes.gtf file in the run or run10x command? Which command, run or run10x, I should use?
What I have tried are described in #320, which mainly got me issue with out-of-memory, but now I got this issue of IndexError: list index out of range.
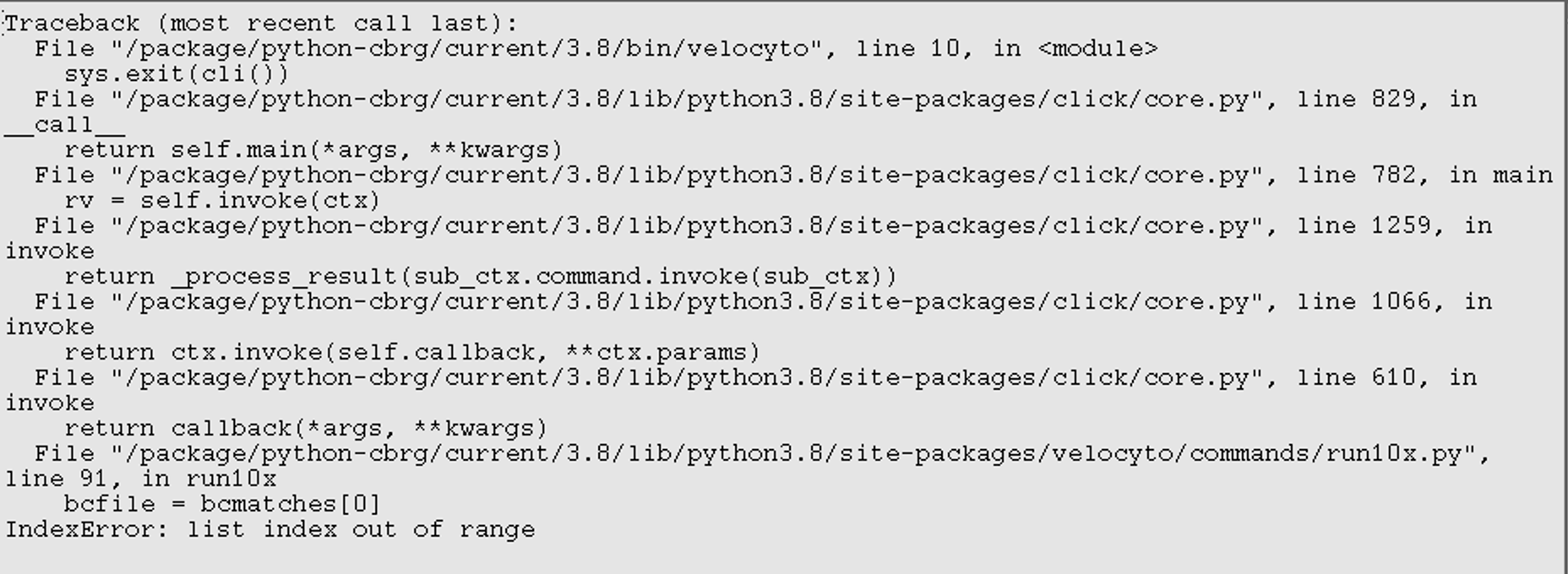
Thank you so much.