tombo
 tombo copied to clipboard
tombo copied to clipboard
raw fraction commonly show many positions with 0.5
Hi there,
I am currently running tombo on a couple of species and what I find is that many of the positions have 0.5 as the raw fraction of reads that are methylated at that position. Any idea why this occurs? my species are commonly homozygous/haploid and therefore it is not a case of haplotype differences
There appear to no errors while running etc, purely the results
I am running this:
tombo detect_modifications alternative_model \ --alternate-bases 5mC 6mA \ --fast5-basedirs fast5_single/ \ --statistics-file-basename 5mC_6mA.tombo \ --processes ${threads} --corrected-group RawGenomeCorrected_001
tombo text_output browser_files \ --statistics-filename 5mC_6mA.tombo.5mC.tombo.stats \ --fast5-basedirs fast5_single/ \ --corrected-group RawGenomeCorrected_001 \ --file-type fraction
Thank you
Is your coverage high everywhere on the genome?
Can you share, say, a histogram showing that 0.5 occurs unusually often?
Have you run these Tombo commands before without running into this issue?
Good luck.
Below is a histogram of the proportion of methylated reads at each position along the genome
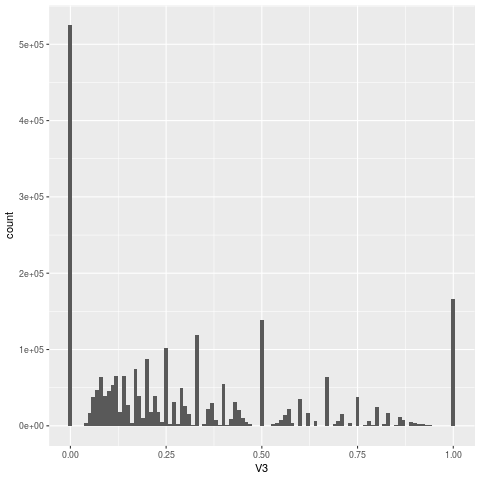
I have this sort of pattern, where the 0.5 occurs just a bit less than the 1 in both Chlamydomonas reinhardtii and multiple strains of Saccharomyces cerevisiae and both are well covered with reads ~40X
I have had this for every run I have done with tombo
Hope that helps
Hi @SAMtoBAM . Thanks for the histogram; that's really helpful.
I'm not sure this is an anomaly. If your coverage is that low (around 40 reads on average) then you should expect that histogram to have peaks where the x-axis is a rational number.
Rational numbers with small numerators and small denominators (in particular 1/2, but also 1/3, 2/3, 3/4, etc) ought to have larger peaks.
I think if you re-run this experiment with a higher coverage depth (maybe 1000x) your histogram will look a lot smoother.