DEP
 DEP copied to clipboard
DEP copied to clipboard
Remove 1 sample and all pvalue adj bump to 1.
Hi DEP's authors,
I have some trouble with the p-value adj. compute by fdrtool.
In my design, I have 7 samples of cond1 and 19 samples of cond2. If I use all these samples, DEP works with 1844 proteins:
- 154 proteins are significant
- the protein x have:
- pvalue: 2.03691168653476E-06
- pvalue adj: 3.32E-13
If I remove 1 sample in my cond2 (7 vs 18), DEP works with 1893:
- no proteins are significant, all pvalue adj bump to 1.
- the protein x have:
- pvalue: 4.14508026818263E-06
- pvalue adj: 1
It doesn't seem to be a bug, but I'm quite surprise that removing one sample change the p-value adj so much! So I'm wondering:
- Why you have choose this method compare to p.adjust() and the qvalue package ?
- If the fdrtool package is appropriate in all case, to calculate a p-value adj ?
If the answer of 2) is not: 3) Have you an idea why my case doesn't work ?
Bonus: is It interesting to offer the possibility to change the method ? (I can work on it).
Thanks for your work and your help, Eric.
Here some fdrtool plots, with all samples:
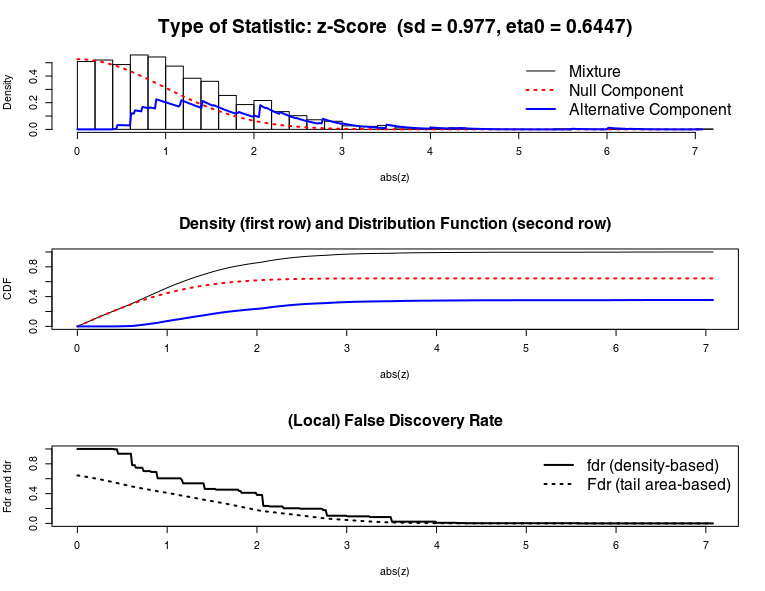
Here some fdrtool plots, after removing 1 sample :
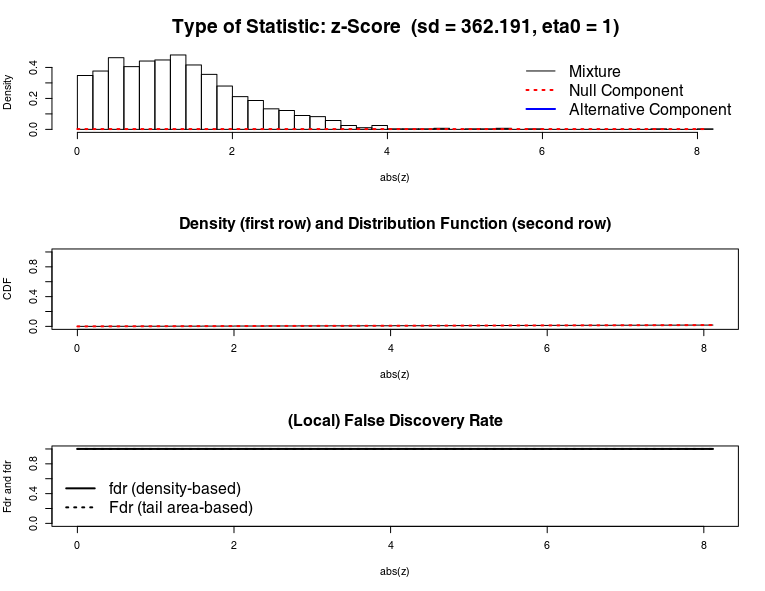
The null component doesn't fit at all the mixture.
In that case, fdrtool (with default param) can't ajust the pvalue. Which is why all pvalue adj bump to 1.
Hi ericloud, I got similar problem as yours when running DEP. I got very few differentially expressed proteins if I use p.adj to filter since most of them are very high. For example, the p value of one protein is 4.8e-5, but the p.adj=0.2. It seems that fdrtool use t-statistic-derived FDRs. I thought maybe I can adjust the p value using BH method by myself using the p values output from DEP. But I am not sure if it is correct way to do. The DEP authors hasn't answer the issues for a long time. Does anyone have any idea? Thx a lot.